儿童和青壮年T细胞淋巴母细胞淋巴瘤的分期、分子特征、风险分层及治疗
时间:2025-09-08 12:15:15 来源 网络 作者:网络
T细胞淋巴母细胞淋巴瘤(T-LBL)是儿童及青少年中第二常见的非霍奇金淋巴瘤(NHL)亚型,与T细胞急性淋巴母细胞白血病(T-ALL)在生物学和临床特征上高度重叠。目前来说,两者主要依据初诊时骨髓(BM)受累程度进行临床区分:若骨髓中原始细胞≥25%,则定义为T-ALL。尽管WHO将T-LBL与T-ALL归入同一疾病亚型,但最新研究提示两者在临床及分子层面存在显著差异。
《British Journal of Haematology》近日发表综述,聚焦儿童T-LBL,系统梳理其临床表现、免疫表型及分子特征;重点探讨年龄相关的基因组模式、复发性突变和染色体异常及其预后意义。值得注意的是,近期研究发现NOTCH1基因融合仅见于儿童T-LBL、在T-ALL中缺失,并与复发风险升高显著相关。
可测量残留病(MRD)在T-LBL中的适用性有限、风险分层尚缺乏统一标准,且亟须针对T-LBL的治疗策略调整。应建立基于生物学特征的风险适应性(risk-adapted)治疗方案(即,根据患者风险调整治疗),并加强协作研究,以改善儿童T-LBL患者的预后与生存质量。

引言
T细胞淋巴母细胞淋巴瘤(T-LBL)约占儿童非霍奇金淋巴瘤(NHL)病例的25%,是一种源于未成熟T细胞的高度侵袭性恶性肿瘤。与急性淋巴母细胞白血病(ALL)以B系基因组异常为主不同,LBL病例携带有驱动恶性转化的突变。在全球范围内,LBL各亚型的分布存在地域差异:与发达地区相比,发展中国家B细胞淋巴瘤比例较低,而T细胞及NK细胞淋巴瘤比例较高。
越来越多的证据提示,T-LBL与T-ALL可能是两种不同的疾病亚型,而非同一疾病在淋巴结、骨髓(BM)和中枢神经系统(CNS)受累倾向上的差异表现。多项研究证实,采用ALL样方案治疗T-LBL可获得极佳疗效,总体生存率超过80%。然而复发性T-LBL的预后极差,长期生存率极少超过20%–30%。目前,针对可靶向分子通路的精准治疗及新型免疫疗法正在积极探索中,有望显著提高复发LBL的挽救成功率。
临床特征与流行病学
本综述中,儿童与青少年定义为年龄<18岁;鉴于文献中定义不一,年轻成人则定义为≤39岁。
T-LBL 的临床表现
根据WHO标准,T-ALL和T-LBL的区分基于骨髓(BM)受累程度:骨髓中淋巴母细胞≥25%定义为白血病,而<25%则定义为淋巴瘤。这一阈值主要通过骨髓穿刺涂片的形态学评估来确定。
儿童队列中T-LBL的中位发病年龄约为8–9岁:患者常表现为纵隔肿块(70%)以及心包或胸腔积液。T-LBL(以及T-ALL)的发病率在儿童期相对稳定,但在婴儿和年轻成人中频率较低。T-LBL(以及T-ALL)以男性为主,男女比例约为2.5:1。这种性别差异几乎在所有年龄组中都存在,但3岁以下儿童除外。已发现X连锁的PHF6肿瘤抑制基因的失活突变和缺失,可能有助于解释这种性别差异。
大约15%–20%的T-LBL患者在初诊时表现为骨髓病变(<25%),与T-ALL不同的是,T-LBL很少累及CNS。这种CNS受累差异的生物学基础尚不清楚,但可能与循环系统原始细胞的数量有关。
如果存在大的纵隔肿块,患者常可出现相关症状,包括呼吸急促、咳嗽和胸痛。上腔静脉(SVC)综合征也很常见,由于上腔静脉血流受阻引起,表现为面部肿胀和发红、颈部静脉扩张、咳嗽和呼吸困难。
对于病情危重的患者,快速稳定疾病至关重要。通常需要在进行诊断程序之前开始使用糖皮质激素,并密切监测和管理肿瘤溶解综合征。由于存在进一步气道压迫、血管受压和急性呼吸衰竭的风险,通常避免在存在大的纵隔肿块时进行麻醉和镇静。如有必要,可以引流心包和胸腔积液,积液可用于诊断性流式细胞术和细胞学评估。
初诊时应进行详细的个人和家族病史询问以及全面的体格检查。一些癌症易感综合征与T-LBL的发生有关,包括Nijmegen breakage综合征(NBS)和遗传性错配修复缺陷综合征(constitutional mismatch repair syndrome,CMMRD)。NBS是由NBN基因的双等位胚系突变引起,该基因编码nibrin蛋白,参与双链DNA断裂的修复。NBS的特征包括小头畸形、身材矮小、免疫缺陷以及易患T-LBL和B细胞非霍奇金淋巴瘤(NHL)。CMMRD是由错配修复基因(MLH1、MSH2、MSH6、PMS2)的双等位致病变异引起的,也易患T-LBL。其临床表现包括咖啡牛奶斑、局部皮肤色素减退、静脉异常和毛基质瘤。如果存在令人担忧的临床特征、Lynch综合征家族史或近亲结婚的情况,应考虑进行胚系基因检测。
分期
过去,T-LBL和B-LBL的分期均采用Murphy(St Jude)分期系统(附表S1)。
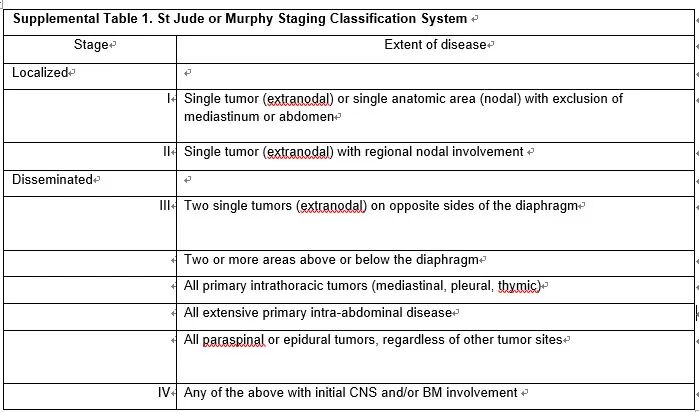
近年来引入了国际儿童非霍奇金淋巴瘤分期系统(International Pediatric Non-Hodgkin Lymphoma Staging System,INPNHLSS)用于更精确的分期(表1)。

完整的疾病分期包括淋巴结、腹部和睾丸的基线超声,以及受累部位的计算机断层扫描 (CT) 或磁共振成像 (MRI);或颈部、胸部、腹部和骨盆的基线CT;双侧 BM 穿刺和活检;以及用于脑脊液细胞计数和细胞学检查的腰椎穿刺。正电子发射断层扫描 (PET) 也更常规地推荐作为基线成像和初始治疗后的随访评价,以评估 PET 高摄取疾病的消退。MRI 可用于颈部、腹部和骨盆,但在肺部疾病评价方面不如 CT 有效。
多参数流式细胞术、聚合酶链反应或二代测序 (NG) 表示基线时的最小播散性疾病 (MDD) 可检测到最小骨髓浸润。这可能在初始化疗区组后重新评估,以确定 BM 微小残留病 (MRD)。
病理学和免疫表型
分类挑战
通过流式细胞术对LBL进行免疫学分类时,遵循欧洲白血病免疫分型协作组(EGIL)标准或WHO标准,以区分T细胞系和前体B细胞系。T-LBL具有特定的定义特征,但EGIL和WHO标准存在显著差异(表2)。WHO指南主要基于对ALL的流式细胞术分析,这些标准无法直接应用于LBL的组织病理学诊断。这种差异给不同诊断平台间对LBL的准确分类带来了挑战。
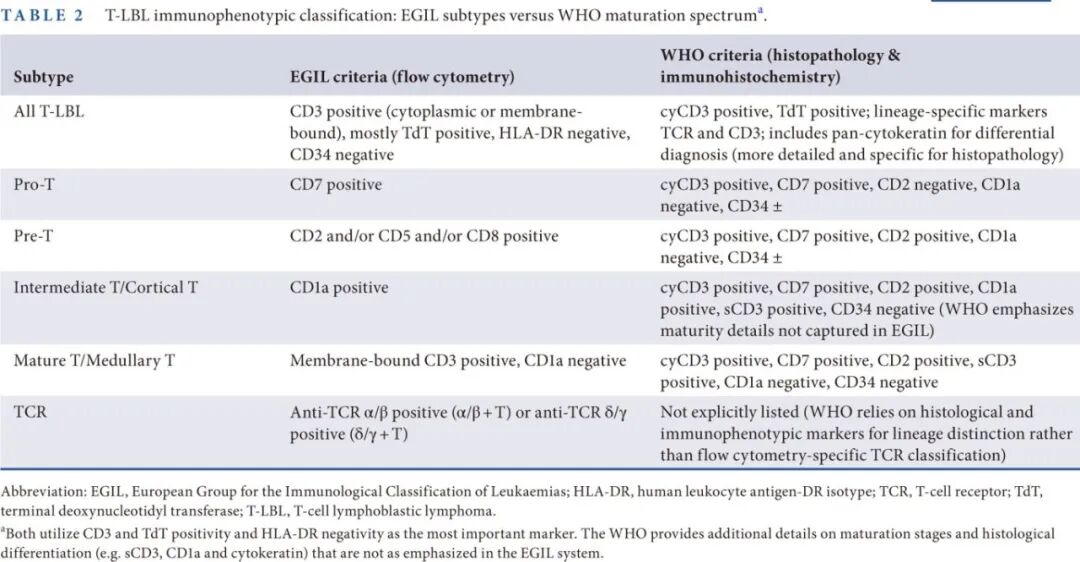
T-LBL的病理特征与免疫表型
T-LBL的特为是中等大小的淋巴母细胞增殖,这些细胞具有圆形至卵圆形的核、细腻的染色质、不明显的核仁以及高核质比。这些原始细胞可浸润胸腺、淋巴结以及胸膜、骨髓和中枢神经系统等结外部位。T-LBL细胞常表达末端脱氧核苷酸转移酶(TdT,一种未成熟T细胞的标记物)和胞质或膜CD3(泛T细胞标记物)。其他T系标记物,如CD1a、CD2、CD4、CD5、CD7和CD8,根据T细胞母细胞的分化阶段而存在表达差异。T-LBL中CD4和CD8的共表达较为常见,提示处于皮质胸腺细胞阶段。
早期T细胞前体(ETP)-LBL约占T-LBL病例的4%,表现为早期分化,表达CD34和髓系标记。这种亚型缺乏常见的CDKN2A/B缺失,ETV6缺失更多见,并携带类似急性髓系白血病(AML)的突变(如FLT3、DNMT3A),而非NOTCH1突变。其预后与非ETP病例相似。
EGIL分类根据表面标记将T-LBL分为四个亚型。相比之下,WHO分类根据成熟阶段定义T-LBL的免疫表型谱系,并指出EGIL曾归为pro-T或pre-T的病例通常符合早期T细胞前体ALL(ETP-ALL)的标准。EGIL免疫表型亚型与WHO定义的成熟谱系的对比总结见表2。
T-LBL的分子特征及预后参数
T-LBL的当前分子发现
在儿童T-LBL的分子遗传学方面,目前仍存在知识空白。已发现T-LBL与T-ALL存在一些遗传学重叠(表3、表4和图1)。
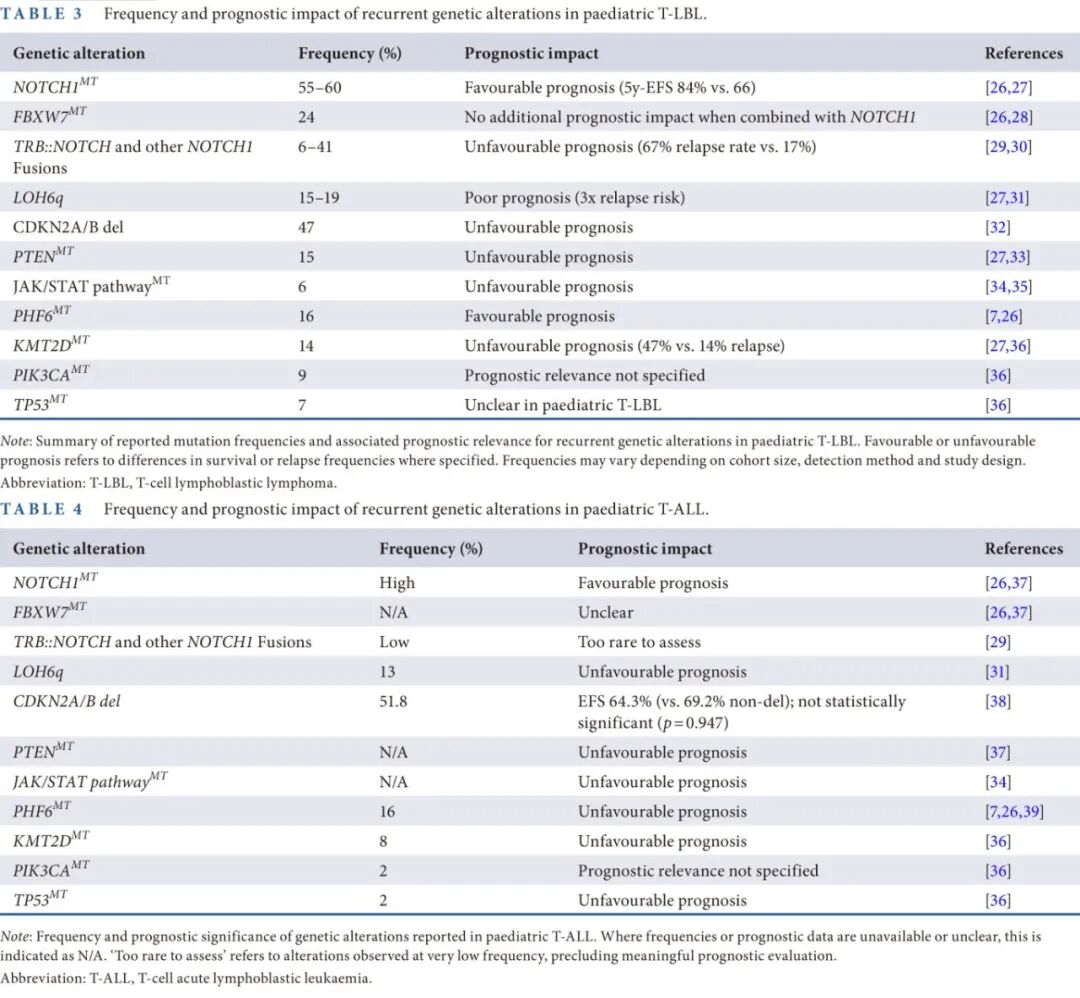
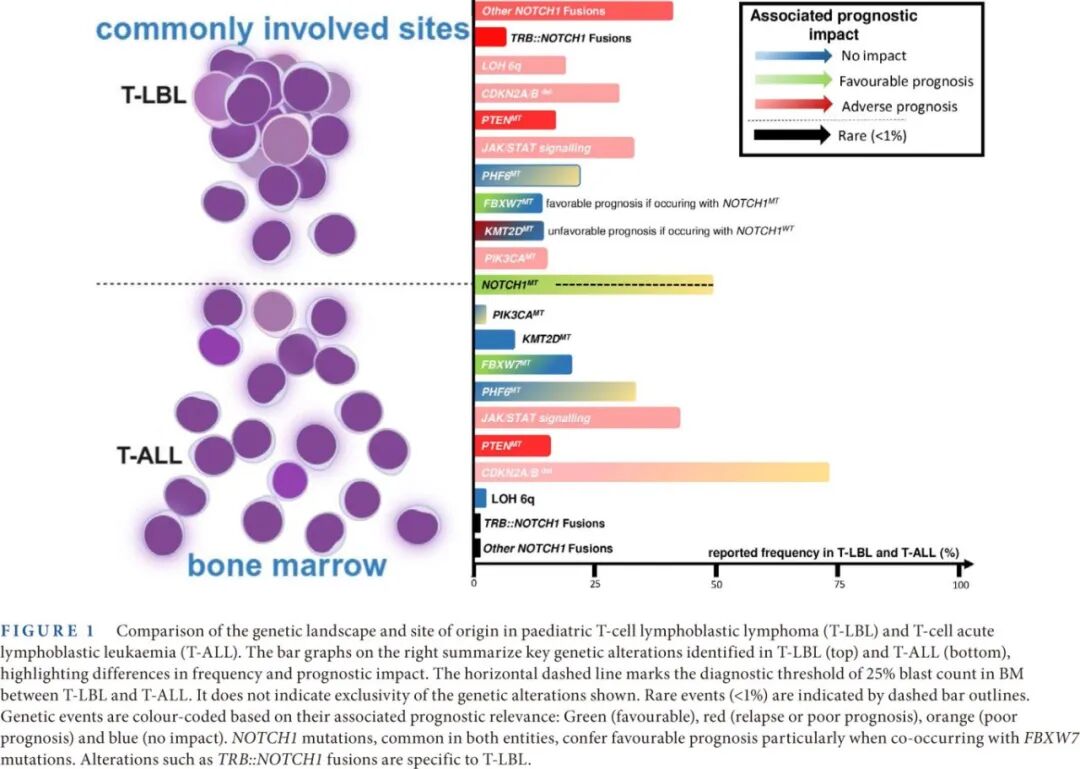
研究已识别出T-LBL的几个新的候选基因,如PAPPA、NFIL3和ZNF91,以及已确立的基因,如NOTCH1、PHF6、MUC4和PRDM2。Bontoux等2022年报告了T-LBL和T-ALL患者之间的差异,例如T-LBL中PIK3CA(30/330;9%)突变的患病率高于T-ALL(8/474;2%),尤其是PIK3CA H1047热点突变。与T-ALL相比,T-LBL中EZH2(43/330;13%)和TP53突变更为频繁。
T-LBL存在6号染色体缺失(LOH6q),且与不良预后相关。三项研究显示,约19%的T-LBL患者检测到LOH6q,涉及的染色体带为6q16。最近有研究显示,约15%的T-LBL病例中存在LOH6q,与复发风险增加三倍相关。在T-LBL中,缺失靶向的基因包括GRIK2、CASP8AP2和EPHA7,但它们的具体作用尚不清楚。
NOTCH1通路突变
NOTCH1和FBXW7突变(N/F突变)调节细胞增殖和分化,其过度激活会促进肿瘤生长。Bontoux等人发现,在包括成人和儿童T-LBL患者的队列中,52%(170/330)存在NOTCH1改变,24%(80/330)存在FBXW7突变。N/F突变在儿童T-LBL病例中发生率约为55% - 60%。配体结合后,全长NOTCH1发生蛋白水解切割,释放出细胞内结构域(ICN1),ICN1转移到细胞核并激活下游靶基因。激活型NOTCH1突变在T-ALL和T-LBL中都很常见,通常影响异二聚体化(HD)结构域和/或PEST结构域,后者通过阻止ICN1降解来稳定ICN1。这些突变与更好的预后相关,如Bonn等2013年的研究显示,存在NOTCH1突变的患者5年无事件生存率(EFS)为84%,而无突变的患者为66%。然而,当FBXW7突变与NOTCH1突变共存时,并未显著改善预后。
miRNA(microRNA)通过调节NOTCH1通路影响T-LBL的生物学特性和预后。miRNA是小的非编码RNA分子,通过结合目标mRNA在转录后水平调节基因表达,例如miR-211、miR-17和miR-223。
NOTCH1易位
30多年前,首次在少数T-ALL病例中描述了NOTCH1与T细胞受体β(TRB)之间的易位,这些易位导致染色体7q34上的TRB位点与染色体9q34.3上的NOTCH1并置。易位断裂点通常发生在NOTCH1的第24外显子之后,使截短的ICN1置于TRB启动子的控制之下,从而导致其配体非依赖性激活。最初的TRB::NOTCH1阳性样本来源于恶性胸腔积液,表明T-LBL中可能比T-ALL中更富集这种易位。
最近,te Vrugt等人(2024年)系统地筛查了一个大型队列(192例T-LBL和167例儿童T-ALL患者),发现在6%的T-LBL病例存在TRB::NOTCH1融合,但在T-ALL患者中没有发现。该易位与显著更高的复发率(67%对比融合阴性T-LBL病例的17%)和较差的EFS相关。
这些发现由Kroeze等人(2024年)独立验证,他们通过RNA测序报告在41%的儿童T-LBL病例中发现NOTCH1融合。值得注意的是,在6例融合阳性患者中,有4例复发且未能存活。te Vrugt等人识别出1例携带N/F突变病例,支持TRB::NOTCH1融合是NOTCH1激活的替代机制的假设。这些融合导致截短的NOTCH1配体非依赖性过表达,绕过生理调节机制。这种机制在生物学上与HD或PEST结构域中的激活突变不同,后者悖论性地与有利的临床结局相关。这种差异的原因尚不清楚,但可能与ICN1剂量和随后的MYC激活水平的差异有关,后者可能对化疗反应有影响。
PI3K-AKT通路突变
PI3K-AKT通路在T-LBL中经常发生改变,在细胞生长和存活中起着核心作用。在T-LBL病例中发现了PIK3CA突变,其发生率显著高于T-ALL(9% vs 2%)。PTEN突变会负向调节该通路,也在15%的T-LBL病例中与不良预后相关。
JAK-STAT和MAPK通路突变
在T-LBL中,STAT5B突变、JAK3突变和IL7R突变等基因突变会激活JAK-STAT通路,这种激活最终会导致治疗抵抗和不良预后。这些突变在大约6%的T-LBL病例中出现,而NRAS突变和KRAS突变则出现在10%的病例中,与T-ALL类似,这些突变并不会对预后产生负面影响。
KMT2D突变
KMT2D突变参与表观遗传调控,在T-LBL中与不良预后相关。携带KMT2D突变的患者往往会有更高的复发率(47% vs 野生型患者的14%)。这些突变常常与PTEN突变共存,当N/F突变缺失时,尤其显著,导致复发率高达67%±15%,相比之下,KMT2D野生型/PTEN野生型的复发率为11%±6%。Bontoux等2022年报告说,在T-LBL中KMT2D突变的发生率(330例中的46例,14%)高于T-ALL(474例中的28例,8%),在儿童和成人病例之间没有显著差异,突变主要聚集在第11外显子,位于2000至3000号氨基酸之间。
TP53突变
在儿童和成人T-LBL中观察到TP53突变的发生率为7%(330例中的22例,对比474例中的9例,2%)。然而由于病例数量较少,TP53突变在儿童T-LBL中的预后意义尚不清楚。
风险分层
T-LBL的风险分层
T-LBL的风险分层仍然具有挑战性。尽管经过广泛评估,但年龄、性别和纵隔肿块均与预后并无相关性。在EURO-LB02研究中,基于分期的风险分层并未显示出EFS的差异;在233例患者中,只有8例为低分期。
对COG A5971研究的回顾性分析发现,67%的患者在诊断时可检测到MDD,并且MDD≥1%与较差的EFS相关。在EURO-LB02中,MDD>1%并无预后影响。在N/F突变患者中,MDD<0.1%并无影响,但在N/F野生型患者中可识别复发风险。
AALL0434将MDD>1%的患者分层为高危治疗,但各风险组之间生存率相似。AALL1231将标危定义为MDD<1%、无中枢神经系统疾病、无类固醇预处理,以及在诱导治疗结束(EOI)时达到≥部分缓解(PR);中危包括在EOI时有任何不利特征且达到≥PR。极高危(VHR)患者具有任何IR临床特征,但在EOI时未达到稳定疾病以上。极高危患者在疾病重新评估前接受了三个block的强化治疗。在AALL1231中,MDD、影像学反应和临床特征并无预后价值,但EOI时MRD<0.1%与更好的EFS相关。单独的诊断性MDD可能并无预后价值,可能需要与分子特征整合。或者,如果在EOI时不使用MDD来提高患者风险并加强治疗,那么在巩固治疗结束(EOC)时重新评估MDD可能仍然有意义,正如即将进行的COG试验所计划的那样。
治疗方案
T-LBL的一线治疗
在过去的几十年中,T-LBL患者的预后显著改善,采用ALL基础治疗方案的EFS达到大约75% - 90%。大多数现代治疗方案都源于两种基础方案之一:纪念斯隆-凯特琳癌症中心的LSA2L2方案,或基于欧洲ALL-BFM方案的NHL-柏林-法兰克福-慕尼黑(BFM)方案。诊断时CNS受累的情况很少见。目前,针对CNS导向治疗的通用方法是按照ALL的标准护理进行经验性鞘内(IT)预防。对于CNS3期疾病的患者,目前的标准做法是进行颅脑放疗。
在AALL0434试验中,对高危T-LBL患者测试了nelarabine但未改善预后,因此并非标准治疗。可能的原因是T-LBL的CNS复发率较低,这一点与T-ALL不同。随后的COG试验AALL1231将T-ALL和T-LBL患者随机分配到改良的增强BFM化疗方案中,在诱导治疗和延迟强化治疗期间,部分患者使用蛋白酶体抑制剂硼替佐米。对AALL0434增强BFM基础方案进行了几项更改,以增强CNS导向治疗。AALL0434在诱导治疗和维持治疗中使用了泼尼松,而AALL1231则在这些周期中使用了地塞米松。总体而言,对于T-LBL患者,硼替佐米改善了4年EFS/OS(OS 90%对比78%),但在T-ALL中对结果并无影响。然而,在诱导治疗和延迟强化治疗中使用地塞米松而非泼尼松与治疗相关死亡率增加有关,特别是在T-LBL患者中;因此,在COG基础方案中,泼尼松仍然是T-LBL的首选类固醇方案。国际试验LBL 2018也在使用NHL-BFM基础方案,并研究在诱导治疗中使用较短的14天地塞米松疗程对比21天泼尼松是否可以减少CNS相关复发(<18岁)。这项研究首次将分子特征(NOTCH1和FBXW7)纳入风险分层。
T-LBL的复发治疗
尽管新诊断LBL患者的预后显著改善,但仍有约10%-20%的患者复发,复发后的预后极差。目前复发T-LBL的唯一治愈方法为造血干细胞移植。再诱导治疗通常包括多药化疗,目标为实现完全缓解(理想情况下MRD阴性)和CNS导向治疗。在欧洲,更倾向于使用强化再诱导方案,而非经典的ALL样的诱导方案。
UKALLR3、ALL-REZ BFM、AALL07P1、DELPHINUS和NECTAR等多种再诱导方案在小规模、非随机的早期试验中进行测试,但T-LBL病例数量较少,因此比较其疗效具有挑战性。方案选择通常基于药物的可及性、之前的毒性、复发时的CNS状态以及最近的化疗暴露情况。
T-LBL的未来方向
正在研究针对复发/难治性T-ALL和T-LBL的新型免疫疗法。尽管CAR-T改变了复发性B系淋巴瘤和白血病的治疗格局,但对于T系疾病存在特定挑战,包括自相残杀、白血病污染、移植物抗宿主病和免疫抑制。一项CD7 CAR-T的I期研究显示,在8名复发/难治性T-LBL成年患者中,有5名在输注后第28天实现髓外疾病CR。6名患者在CAR-T治疗后接受了异基因HSCT;其中1名死于GVHD。在最近一次随访时间点(中位数98.5天,范围55 - 166天),4名患者仍处于髓外缓解状态(1名患者在CAR-T治疗前因先前接受过放疗而无髓外疾病)。
T-LBL还需要其他免疫疗法和精准医疗方法,但由于病例数量较少且需要在诊断时迅速开始治疗,分子特征分析一直具有挑战性。Hem-iSMART(国际概念验证治疗分层试验,针对儿童复发或难治性血液系统恶性肿瘤的分子异常)是一项国际多中心I/II期平台试验,目标是针对复发/难治性T-ALL和T-LBL的儿童、青少年和年轻成人,检验多种药物和靶向疗法。基于入组时进行的分子分析和药物敏感性测试,患者被分配到几个子研究组。结果可能对未来基于精准的挽救疗法具有启发性。
总结
根据基于BFM的ALL式治疗方案,儿童、青少年和年轻成人T-LBL患者的生存预后在过去几十年中有所改善。幸存者因治疗而遭受许多长期不良影响,复发后的预后极差。在ALL中已出现了有关免疫疗法获益的良好数据,但尚不清楚淋巴瘤是否会看到同样的影响。包括更多T-LBL患者在内的国际协作努力,将有助于了解免疫疗法和基因组指导的精准医疗方法,目标是防止更多患者复发并减轻毒性。鉴于越来越多的证据表明LBL和ALL在临床表现、分子特征、治疗反应和复发生物学方面存在差异,应开发针对疾病的特定治疗策略。未来方案还应将LBL特定的危险因素纳入不同的分层和治疗策略中。
参考文献
te Vrugt M, Newman H, Teachey DT, Burkhardt B. T-cell lymphoblastic lymphoma in children, adolescents and young adults. Br J Haematol. 2025;00:1–9. https://doi.org/10.1111/ bjh.70053

