前言
内质网(ER)是分泌和跨膜蛋白合成、折叠和修饰的中心细胞器。ER中的蛋白质处理、修饰和折叠是决定细胞功能、命运和生存的严格调控过程。在几种肿瘤类型中,不同的致癌、转录和代谢异常协同作用,产生不利的微环境,破坏肿瘤细胞和间质细胞的内质网稳态,以及浸润淋巴细胞。
这些变化引发了持续的内质网应激状态,已经证明这种应激状态控制着癌细胞的多种促肿瘤属性,同时动态地重新编程固有免疫细胞和适应性免疫细胞的功能。ER应激传感器及其下游信号通路的异常激活已成为肿瘤生长和转移以及对化疗、靶向治疗和免疫治疗反应的关键调节因子。
TME中ER应激的常见驱动因素
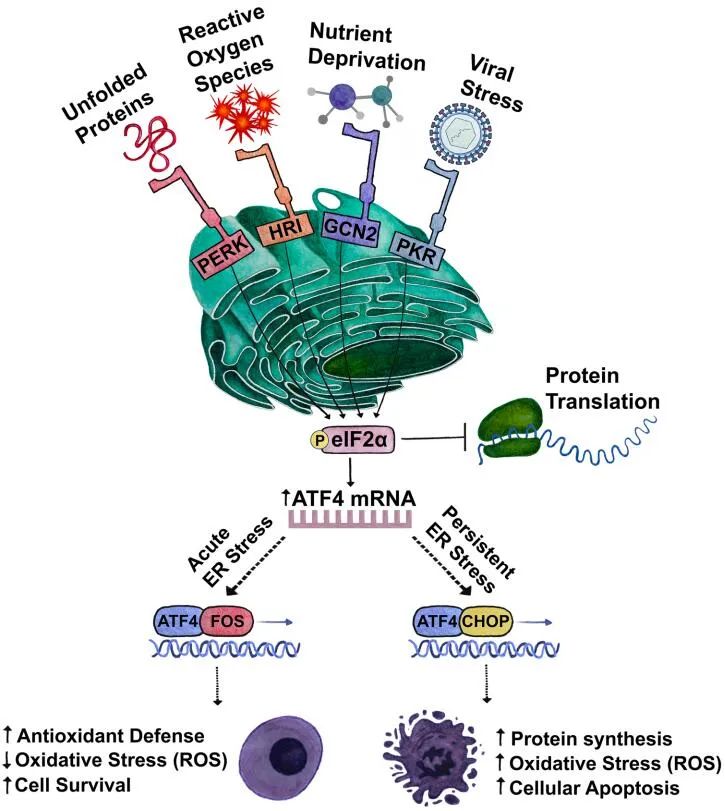
内质网蛋白质折叠和修饰是一个高度调控的过程。然而,各种内源性和外源性应激可以破坏细胞器中的蛋白质稳态,导致内质网应激。未折叠蛋白反应(UPR)是一种高度保守的适应性机制,有三个分支协调对未折叠或错误折叠蛋白质的有害累积的反应。它们包括:需肌醇酶1α(IRE1α)、类PRKR ER激酶(PERK)和激活转录因子6(AFT6)。
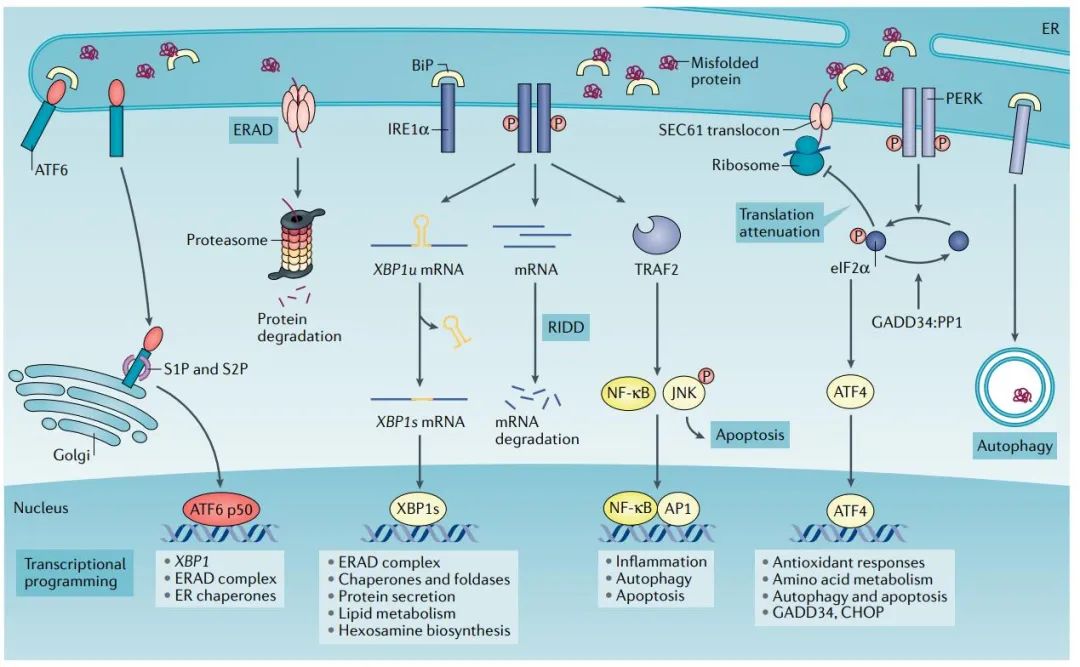
TME中丰富的多种应激源动态地干扰了恶性细胞和间质细胞内质网的蛋白质折叠能力。
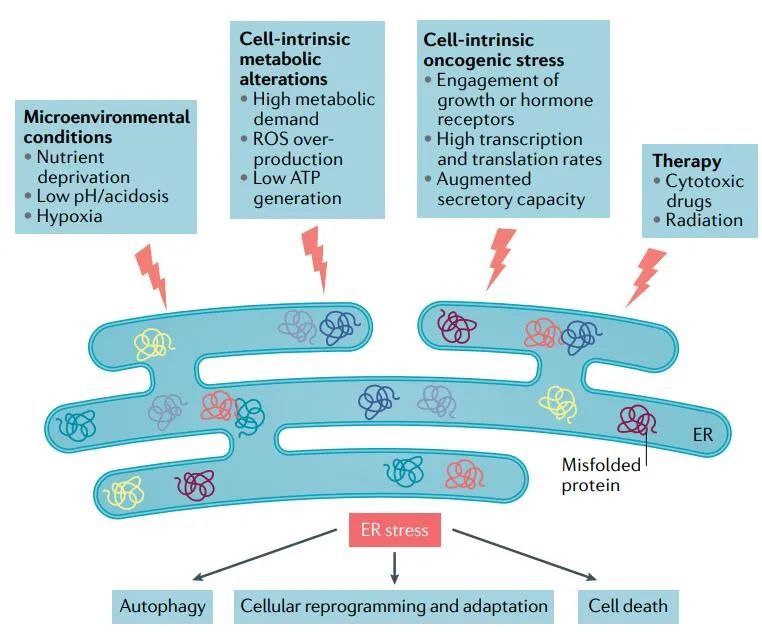
缺氧:缺氧是TME的一个常见特征,它扰乱内质网的稳态。蛋白质合成过程中二硫键的形成可以在没有氧气的情况下发生,但翻译后折叠或异构化是氧依赖的过程。此外,缺氧限制了氧依赖性内质网局部氧化还原酶ERO1α的功能,这是形成二硫键和蛋白质折叠所必需的。
营养:与正常能量需求相比,代谢应激以营养供应不足或过量为特征,很容易破坏内质网的稳态。葡萄糖和谷氨酰胺的有效性与内质网应激密切相关。
活性氧:内质网中的蛋白质折叠很大程度上依赖于这个细胞器的氧化还原状态。细胞内活性氧(ROS)的积累对外界条件的反应或由不同的信号事件引起,可以极大地扰乱内质网蛋白表达。
低pH:癌细胞利用有氧糖酵解作为中心代谢途径,从而产生乳酸,降低周围微环境的pH值。
肿瘤细胞内质网应激反应
UPR在肿瘤转化和肿瘤生长中的作用
致癌转化是一个多步骤的过程,它利用UPR来克服各种障碍。正常上皮细胞中MYC的过度激活会产生大量的蛋白毒性应激,并导致细胞存活率下降。然而,在包括淋巴瘤、神经母细胞瘤、前列腺癌和乳腺癌在内的多种人类癌症中,经受MYC诱导应激的细胞表现出UPR活性的增强。因此,完全激活的UPR对于适应由MYC驱动的癌基因转化引起的压力至关重要。
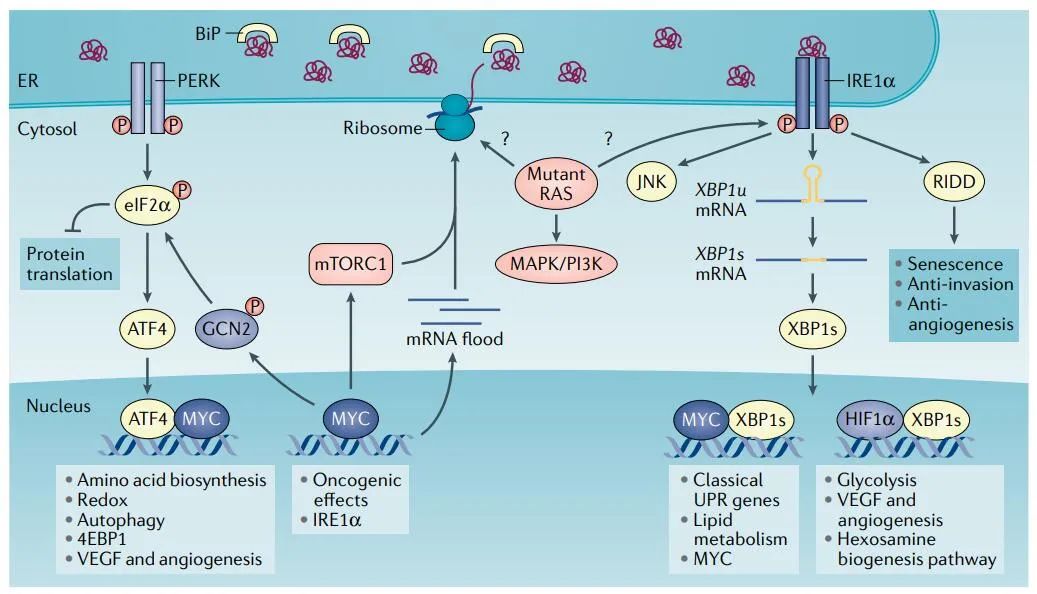
突变体RAS是另一个与UPR相互作用的致癌驱动因素。获得性耐药使KRAS突变型肺癌细胞能够绕过典型的KRAS效应器,但需要过度活跃的AXL/eIF4E,增加ER中的蛋白质周转,以及ER应激缓解UPR生存途径的适应性激活,HSP90维持其完整性。HSP90抑制剂可以协同增强MTA和trametinib的抗肿瘤作用。
分子伴侣结合免疫球蛋白(BiP)在多种人类癌症中过度表达,并通过多种机制促进肿瘤生长,如促进生长因子成熟和分泌、抑制细胞凋亡和促进血管生成。因此,BiP被认为是治疗人类癌症的一个有吸引力的靶点。
PERK利用各种机制来调节肿瘤的进展。首先,它的一个关键功能是通过增加抗氧化剂谷胱甘肽的生物合成来控制氧化应激。第二,PERK–eIF2α轴减弱整体转录并增强自噬作用,以促进MYC驱动的淋巴瘤中的细胞保护性UPR功能。第三,PERK和GCN2都被MYC激活以磷酸化eIF2α并诱导ATF4。
UPR在转移和休眠中的作用
一些研究表明PERK–eIF2α轴抑制失巢,是肿瘤侵袭和转移所必需的。PERK在经历上皮-间充质转化(EMT)的细胞中也被选择性激活,其分泌能力增强。在原发性肿瘤中,转移细胞在循环和远处组织中的氧化应激水平高于癌细胞。代谢适应,如抗氧化剂的合成,对于癌细胞在远处的生存和最终的生长是必不可少的。PERK分支通过ATF4和NRF2促进抗氧化反应,因此,可能通过减轻氧化应激而有益于转移细胞。
最近的研究还表明,来自病人和小鼠癌症模型的休眠恶性细胞的UPR反应增加,包括乳腺癌、鳞癌、结直肠癌和胰腺导管腺癌(PDAC)。UPR可能诱导休眠,作为在远端器官的不利微环境中生存的一种适应。
肿瘤细胞ER应激对肿瘤免疫微环境的调节
大量研究表明,肿瘤细胞内在的ER应激反应可以通过改变共存于TME中的免疫细胞的功能来影响肿瘤进展。
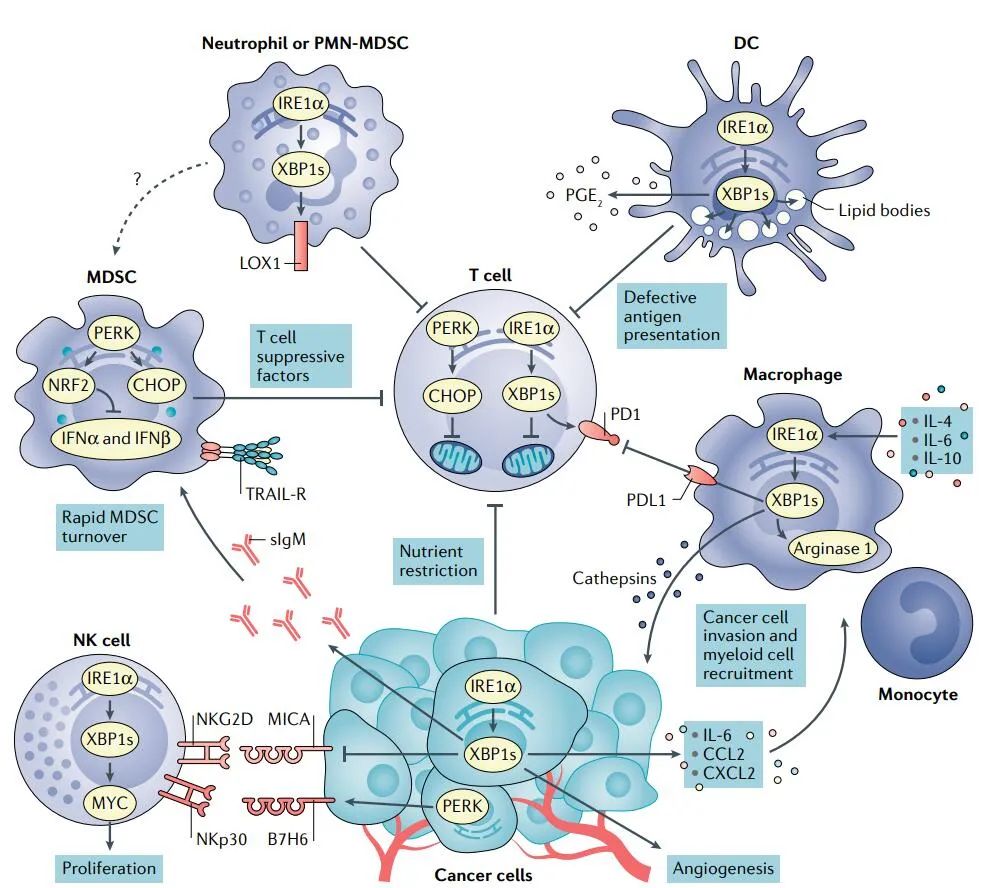
早期研究表明,ER应激的诱导和UPR的激活可能通过XBP1s和ATF6过度表达抑制主要组织相容性复合体I类(MHC-I)分子的表面表达。
肿瘤细胞ER应激反应被认为可以改变自然杀伤细胞(NK)介导的肿瘤识别。研究表明,在经历药物性ER应激的黑色素瘤细胞中,激活UPR的PERK–eIF2α轴可以诱导B7H6的表达,B7H6是NK细胞受体NKp30的配体。
ER应激的肿瘤细胞可以释放额外的因子来招募或改变肿瘤中髓系细胞的功能。
肿瘤细胞ER应激还可以调节T细胞介导的肿瘤生长、转移和对免疫治疗的反应。在接受抗CTLA-4治疗的各种黑色素瘤患者队列中,XBP1s、ATF4和BiP的表达减少与改善疗效和延长生存期相关。
肿瘤内免疫细胞的UPR
肿瘤细胞的高代谢需求和无限制的增殖能力极大地改变了肿瘤环境的营养成分,而肿瘤浸润性免疫细胞对蛋白质折叠和产生有效抗癌反应所需的关键营养素的获取有限。因此,肿瘤浸润的淋巴细胞持续激活ER应激反应,除了触发典型的UPR外,还以免疫细胞特异的方式调节主要的转录和代谢程序。
靶向内质网应激传感器或其相关的UPR反应途径,可能有助于增强免疫检查点阻断和过继性T细胞免疫治疗对目前这些方法难以治疗的实体瘤的效果。
靶向UPR的药物研究
将标准疗法与UPR调节剂相结合的方法已在临床前癌症模型中显示出显著的疗效,因此值得在癌症患者中进一步研究。
IRE1α抑制剂:IRE1α有两个可靶向的酶结构域:激酶结构域和内啡肽酶结构域。IRE1α激酶抑制剂在多发性骨髓瘤异种移植模型中显示出显著的体内疗效。IRE1α激酶抑制剂KIRA8或AMG-18,抑制多发性骨髓瘤生长,并增强这些肿瘤对已建立的一线药物,蛋白酶体抑制剂硼替佐米和免疫调节药物来那度胺的反应。
IRE1αRNase抑制剂,包括B-I09、STF083010、MKC3946和MKC8866,已在乳腺癌、前列腺癌、黑色素瘤、淋巴瘤、多发性骨髓瘤和CLL的小鼠模型中进行了广泛的试验。B-I09已被证明是一种安全和选择性的IRE1αRNase抑制剂,适合在体内使用。B-I09抑制CLL小鼠模型中的白血病生长而不引起全身毒性,并与FDA批准的Bruton酪氨酸激酶(BTK)抑制剂ibrutinib协同作用,诱导B细胞白血病、淋巴瘤和多发性骨髓瘤的人类细胞系的凋亡。
PERK抑制剂:PERK抑制剂GSK2606414和GSK2656157抑制不同癌症的人类异种移植模型中的肿瘤生长。GSK2656157还可使结肠癌细胞对5-氟尿嘧啶(5-FU)化疗药物敏感。有趣的是,在免疫原性肉瘤小鼠模型中,GSK2606414可激活T细胞功能并增强对PD-1阻断的反应。尽管疗效显著,但PERK抑制对胰腺产生严重的毒性作用,并显著抑制胰岛素的生成。
基于结构的设计和优化使化合物AMG44和AMG52被认为是有效和高度选择性的PERK抑制剂。这两种化合物在体内具有良好的药代动力学特性和耐受性,需要进一步的临床前和临床研究来评估它们与细胞毒性药物或靶向治疗相结合的抗肿瘤疗效和潜在副作用。
eIF2α抑制剂:ISRIB是一种有效的eIF2α抑制剂,通过激活eIF2B来抑制eIF2α磷酸化的影响。当作为一种单一的药物使用时,ISRIB能显著抑制PTEN缺陷和MYC过度表达的前列腺癌进展,延长带瘤小鼠的生存期。重要的是,单用ISRIB可在治疗3周后导致晚期前列腺肿瘤消退,而无明显副作用。
BiP抑制剂:在各种小鼠肿瘤模型中,BiP抑制剂KP1339诱导大范围ER应激和免疫原性细胞死亡。KP1339的I期研究显示,38例转移性神经内分泌肿瘤、非小细胞肺癌(NSCLC)或结肠癌患者中有10例疾病稳定,并且具有可接受的耐受性。
HA15是另一种以BiP为靶点的化合物,在黑色素瘤的异种移植模型中,HA15诱导了显著的抗肿瘤作用,并且有效地减缓了BRAF抑制剂耐药的黑色素瘤,没有主要的副作用。
展望
持续性内质网应激是肿瘤的一个新特征,它是由TME中的多种代谢和致癌异常因素引起的,这些异常扰乱了肿瘤细胞和浸润性免疫细胞的蛋白质折叠稳态。活跃的内质网应激反应使肿瘤细胞适应致癌和环境挑战,同时协调不同的免疫调节机制,促进肿瘤进展。
以内质网应激反应为靶点可以在增强抗肿瘤免疫的同时破坏肿瘤细胞的某些攻击性特质。越来越多的证据表明,对内质网应激传感器或UPR反应的调节,会使侵袭性肿瘤对细胞毒性药物、靶向治疗和免疫治疗更加敏感。更大规模的临床前研究以及对临床试验样本的回顾性分析,有助于发现有效的UPR靶向联合治疗,以获得防止癌症进展和/或复发的持久反应。
参考文献:
1.Endoplasmic reticulum stress signals in the tumour and itsmicroenvironment. Nat Rev Cancer. 2020 Nov 19.
2. HSP90/AXL/eIF4E-regulated unfolded protein response as an acquiredvulnerability in drug-resistant KRAS-mutant lung cancer. Oncogenesis. 2019 Sep; 8(9): 45.
3. Stress relief for cancer immunotherapy: implications for the ERstress response in tumor immunity. Cancer Immunol Immunother. 2020 Oct 26.