引言
在面对病毒、细菌等病原体侵袭以及癌症等疾病时,免疫系统的第一道防线——先天免疫系统发挥着至关重要的作用。然而,免疫信号的失调可能带来严重后果:过于微弱的信号会导致病原体清除不力,而过度活跃的信号则可能引发自身免疫疾病和慢性炎症。因此,了解免疫信号的调控机制是研究人员长期关注的焦点。然而,尽管哺乳动物的免疫信号通路在进化中具有高度保守性,不同物种之间的免疫反应却展现出显著差异,这提示可能存在特定于物种的调控机制,而这些机制的具体分子基础尚不清楚。
12月12日Cell的研究报道“Regulation of human interferon signaling by transposon exonization”,揭示了一个全新的调控机制:转座子(Transposable elements, TEs)的外显子化(exonization)如何通过生成新的蛋白质异构体(isoforms)来调控人类免疫信号。转座子是基因组中的“跳跃基因”,占据了人类基因组超过50%的比例。传统上,转座子被视为基因组中的“寄生者”,但近年来的研究发现,它们能够作为基因调控的重要“工具箱”,参与基因表达的调控。而这项研究进一步发现,某些转座子在免疫基因中的外显子化事件不仅生成了功能性的蛋白质异构体,还在调控免疫信号中发挥了关键作用。
这项研究聚焦于干扰素受体IFNAR2的转座子来源异构体IFNAR2-S,这一异构体通过“充当诱饵受体”(decoy receptor)的方式,在细胞中广泛表达,能够显著抑制干扰素信号的活性。这一机制不仅对理解人类免疫系统的调控提供了新视角,还可能对多种免疫相关疾病(如新冠肺炎、系统性红斑狼疮)和治疗策略带来重要启示。
这项研究的重要性在于,它展示了转座子作为一种进化驱动力如何塑造物种特异性的免疫功能,并揭示了一个调控免疫信号的潜在靶点,为深入探索人类免疫调控和疾病治疗开辟了新的道路。

免疫信号的微妙平衡:保护与危险的双刃剑
在体内,先天免疫系统是抵御外来病原体和清除受损细胞的第一道防线。它通过一系列复杂的信号通路快速识别并反应,对维持健康至关重要。尤其是干扰素(Interferon, IFN)信号作为先天免疫的核心环节,其作用如同一道“警报系统”,能迅速激活抗病毒机制和免疫细胞。然而,这一强大的保护机制背后也潜藏着风险:一旦调控失衡,干扰素信号可能从“卫士”转变为“刽子手”。
当干扰素信号不足时,免疫系统对感染的清除能力大打折扣,这常见于慢性病毒感染和某些免疫缺陷疾病中。例如,在严重的病毒感染中,如新冠肺炎(COVID-19),患者往往因干扰素反应迟钝而错失对病毒的早期控制。反之,干扰素信号过度活跃则会导致自身免疫性疾病和慢性炎症,比如系统性红斑狼疮(Systemic Lupus Erythematosus, SLE),其特征便是“干扰素相关基因”的异常高表达。这种过激反应不仅损伤健康细胞,还可能引发器官功能紊乱。
研究表明,免疫信号的调控需要极为精准,而这背后的复杂性超乎想象。虽然干扰素信号的基本通路在哺乳动物中具有高度保守性,但不同物种之间表现出的免疫反应却存在显著差异。究其原因,研究人员发现,调控这些差异的机制往往特定于物种。该研究揭示了灵长类特有的干扰素受体异构体IFNAR2-S,它通过“诱饵受体”的方式抑制干扰素信号,从而在免疫反应中扮演着“刹车”的角色。这种独特的调控机制帮助灵长类动物在强大免疫防御与避免自身损伤之间找到了平衡。
在这个双刃剑般的系统中,如何精准调控免疫信号,既避免病原体的“漏网之鱼”,又防止自身组织的“误伤”,是免疫学研究的核心问题。而这项研究无疑为我们揭开了这一复杂问题的部分答案。
转座子:从基因组寄生者到调控新星
在人类基因组的浩瀚“星空”中,有一类被称为转座子(Transposable Elements, TEs)的基因片段曾经被视为“基因组寄生者”。这些片段占据了人类基因组超过50%的比例,具备自主“移动”的能力——它们可以通过复制或剪切粘贴的方式在基因组中跳跃,改变自身的位置。长期以来,转座子因其扰乱基因组稳定性而被认为是“不速之客”。然而,随着技术的进步,人们开始意识到,这些“寄生者”并非毫无价值;相反,它们可能是推动基因组进化和功能创新的重要动力之一。
转座子如何从“破坏者”变为“调控者”?答案在于它们的外显子化(exonization)过程。外显子化指的是转座子通过整合到基因的内含子区域,并被细胞的剪接机制(splicing machinery)识别为外显子,从而成为基因表达的组成部分。这一过程使转座子能够产生功能性蛋白质异构体,对基因的表达和调控产生深远影响。
该研究揭示,转座子通过外显子化在免疫基因中扮演了重要角色。研究人员利用长读长转录组测序技术,发现了超过3500个转座子外显子化事件,其中有许多涉及关键免疫基因。这些事件产生的异构体不仅具有翻译潜力,还能够显著影响蛋白质功能。例如,IFNAR2基因的一段灵长类特有的Alu转座子通过外显子化生成了一个独特的异构体IFNAR2-S。与传统的IFNAR2异构体相比,IFNAR2-S缺失了细胞内信号转导结构域,却能作为一种“诱饵受体”有效抑制干扰素信号,从而调控免疫反应的强度。
这一发现将转座子从“基因组垃圾”的地位推向了基因调控的前沿。转座子不仅是一种进化的工具箱,更是细胞通过改造基因组“原料”适应复杂环境的创新策略。
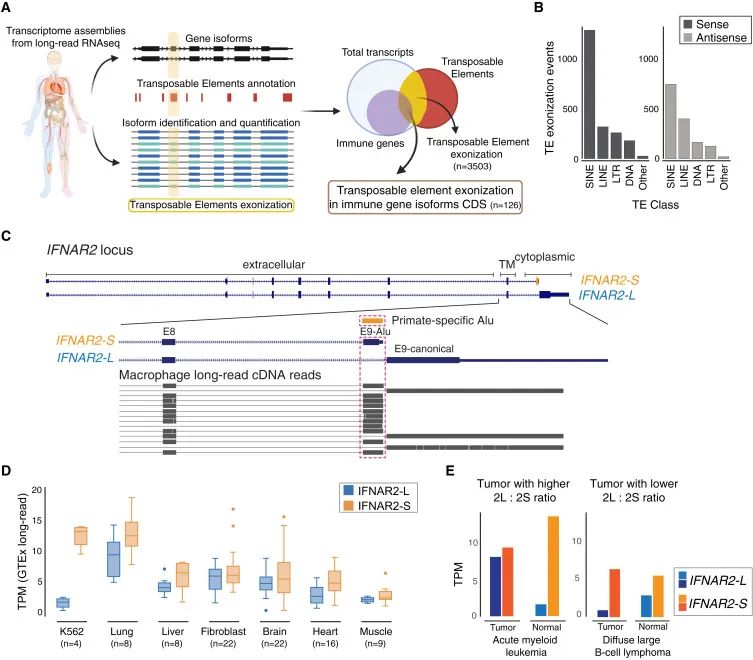
IFNAR2-S和IFNAR2-L的生成、表达及其在免疫调控和疾病中的特异性分布(Credit: Cell)
识别免疫基因中转座子外显子化的策略(图A)
研究采用长读长转录组数据分析技术,筛选了包含转座子外显子的蛋白质编码基因异构体。这些异构体需要满足以下条件:转座子与蛋白质编码基因外显子重叠超过80%,并且异构体的表达水平达到TPM(每百万转录本中的转录本数)≥ 5。该方法为发现转座子如何通过外显子化参与免疫基因调控提供了强有力的支持。
不同类别转座子的外显子化事件统计(图B)
数据统计了各种转座子(TEs)类型的外显子化事件,主要包括SINE(短散布核元件,如Alu)、LINE(长散布核元件)、LTR(长末端重复序列)以及DNA转座子等。研究发现,在免疫基因中,Alu等SINE转座子的外显子化事件最多,且它们可以与母基因的正义链或反义链结合,从而形成新的剪接位点。
IFNAR2-S和IFNAR2-L的异构体比对分析(图C)
在人类巨噬细胞的转录组数据中,研究比对了IFNAR2-S和IFNAR2-L两种异构体的表达。结果显示,两种异构体均被成功检测,表明它们在巨噬细胞中都有活跃的表达。
IFNAR2-S和IFNAR2-L在健康组织中的表达水平(图D)
通过GTEx项目的长读长RNA测序数据,研究分析了健康人类组织中两种异构体的表达分布。结果表明,IFNAR2-S(橙色)的表达水平在多数组织中与IFNAR2-L(蓝色)相当甚至更高。数据以箱线图呈现,显示了每种组织中异构体表达的四分位分布、极值和离群值。
IFNAR2异构体在肿瘤中的表达差异(图E)
结合TCGA数据,研究比较了急性髓系白血病(AML)和弥漫性大B细胞淋巴瘤(DLBCL)中IFNAR2-S和IFNAR2-L在正常组织和癌症组织中的表达水平。结果发现,在某些肿瘤样本中,IFNAR2-S和IFNAR2-L的表达比例发生了显著变化,这提示两种异构体在免疫信号调控中的作用可能随病理状态而动态调整。
独特的“诱饵”:干扰素受体IFNAR2-S的发现
IFNAR2-S的发现源于对人类基因组的深入分析。通过外显子化,这段Alu转座子被纳入了IFNAR2的转录本中,生成了一个新的异构体IFNAR2-S。与传统的全长异构体IFNAR2-L相比,IFNAR2-S缺失了细胞内信号转导所需的关键结构域,仅保留细胞外与干扰素结合的部分。
正因如此,IFNAR2-S无法激活干扰素信号通路中的JAK/STAT级联反应,但它却能够高效地与干扰素结合,从而“诱捕”信号分子,阻止其进一步激活IFNAR2-L。这种“诱饵受体”机制使IFNAR2-S成为干扰素信号的负调控因子,对维持免疫信号的适当强度起到了关键作用。
该研究进一步揭示了IFNAR2-S在组织中的广泛表达及其高表达水平。研究人员发现,在多种正常组织和癌症组织中,IFNAR2-S的表达水平与IFNAR2-L相当甚至更高。这一发现挑战了以往认为IFNAR2-L是主导异构体的传统观点。此外,IFNAR2-S的表达比例在某些疾病状态下会发生显著变化,例如在急性髓系白血病中,IFNAR2-S的比例降低,可能与异常免疫调控相关。
IFNAR2-S的形成机制和独特功能表明,这一异构体是灵长类进化中的重要创新,它通过一种巧妙的方式平衡了干扰素信号的强度。这不仅有助于保护机体免受过度免疫反应的伤害,还为研究疾病中的免疫调控失衡提供了新方向。
免疫的“开关”:IFNAR2-S如何抑制干扰素信号
与传统的全长异构体IFNAR2-L相比,IFNAR2-S缺少细胞内的信号转导结构域,无法激活下游的JAK/STAT通路。但它保留了细胞外的干扰素结合能力。当干扰素(如IFN-β)存在时,IFNAR2-S可以与其紧密结合,并与另一关键受体IFNAR1形成一个“不完全信号复合体”。由于IFNAR2-S无法完成信号传递,这一复合体实际上“锁定”了干扰素分子,阻止它们进一步激活IFNAR2-L及下游信号通路。
在IFNAR2-S缺失的细胞中,干扰素信号的响应显著增强,表现为JAK/STAT级联反应的过度活跃,以及干扰素刺激基因(ISGs)如OASL和ISG15的表达水平显著升高。此外,IFNAR2-S的抑制作用在低浓度干扰素下尤为明显。这表明,IFNAR2-S可以通过降低细胞对干扰素的敏感性来“调低”免疫响应的强度,从而有效控制炎症和组织损伤。
有趣的是,IFNAR2-S还与细胞的受体内吞机制有关。研究发现,IFNAR2-S结合的干扰素并不会像IFNAR2-L那样被迅速内化并降解,而是可能滞留于细胞膜表面,进一步限制干扰素的利用率。这种机制为其发挥“诱饵受体”作用提供了更强的分子基础。
通过精确地调控干扰素信号的强度,IFNAR2-S帮助机体在免疫防御与组织保护之间实现了平衡。
从新冠病毒到癌症:IFNAR2-S在疾病中的潜在作用
IFNAR2-S,这一灵长类特有的“诱饵受体”,在多种病理状态中展现出了其独特的调控作用。无论是面对新冠病毒(SARS-CoV-2)的侵袭,还是肿瘤微环境中的复杂免疫反应,IFNAR2-S对干扰素信号的调控能力为我们揭示了其潜在的临床意义。
在抗病毒免疫中的作用
新冠病毒感染中的免疫反应通常表现为干扰素信号的紊乱。研究表明,IFNAR2-S通过抑制干扰素信号,可能在病毒感染早期帮助限制过度的免疫炎症反应。然而,这种抑制也可能削弱宿主对病毒的抗击能力。在SARS-CoV-2感染的人类细胞模型中,缺失IFNAR2-S的细胞对干扰素β的响应显著增强,不仅下游的干扰素刺激基因(如OASL和ISG15)表达水平更高,抗病毒能力也更强。这种现象提示,在过度免疫反应引发组织损伤和病毒快速复制之间,IFNAR2-S可能扮演了“中和剂”的角色。然而,对于部分携带特定基因变异的个体,例如IFNAR2-S表达比例异常高的人群,可能面临更大的感染风险,因为其免疫反应可能被过度抑制。
在癌症中的表现
在肿瘤微环境中,免疫系统的作用更为复杂。数据揭示,IFNAR2-S和IFNAR2-L在肿瘤中的表达比例会发生显著变化。例如,在急性髓系白血病(AML)中,IFNAR2-S的表达水平降低,可能与干扰素信号增强和异常免疫活性有关。而在某些癌症中,如弥漫性大B细胞淋巴瘤(DLBCL),IFNAR2-S的高表达可能抑制了免疫系统对肿瘤的攻击,使得肿瘤细胞得以逃避免疫监视。这种机制可能为癌症的免疫治疗提供一个新靶点,即通过调节IFNAR2-S的表达比例来增强免疫反应。
无论是抗病毒治疗还是癌症免疫疗法,IFNAR2-S的调控作用都为研究者提供了新的方向。通过靶向调节IFNAR2-S的功能,可能实现对过度炎症或免疫逃逸的精准干预。
进化的礼物:为何IFNAR2-S只存在于灵长类动物?
在免疫系统的复杂演化过程中,一些独特的分子特征成为特定物种的“专属利器”。干扰素受体IFNAR2-S的发现正是这一现象的鲜活例证。它不仅是灵长类动物进化的礼物,也揭示了免疫调控如何因环境和物种需求的变化而塑造出独特的适应策略。
起源于转座子的“神来之笔”
研究显示,IFNAR2-S的形成源于一种名为Alu的转座子。大约在6000万到7000万年前,Alu转座子通过“基因转换”事件被插入到IFNAR2基因的最后一个内含子中。这一事件只发生在灵长类动物的祖先中,而其他哺乳动物中并没有类似的转座子整合。这段Alu序列通过外显子化,成为了IFNAR2-S的编码部分,为干扰素信号调控增添了一种全新的工具。研究数据进一步显示,这一外显子化事件所涉及的关键序列在灵长类动物中具有高度保守性,表明其对免疫功能的重要性。
灵长类动物的独特需求
为何这种调控机制只存在于灵长类动物?答案可能与它们复杂的生态环境和社会行为有关。灵长类动物面临多样化的病原体和频繁的群体接触,这对免疫系统提出了双重挑战:既要提供强大的抗感染能力,又要避免因过度免疫反应造成的组织损伤。IFNAR2-S的“诱饵受体”机制,能够灵活调节免疫信号的强度,既保证了抗感染的基础防御,又降低了因过激免疫反应导致自身组织受损的风险。
进化与功能的精妙平衡
IFNAR2-S的进化不仅是一个巧合,也是物种特异性免疫调控的必然产物。这一“礼物”展现了转座子如何作为进化的“工具箱”,为灵长类动物提供了一种精妙的免疫调节手段。这种特异性机制不仅是灵长类免疫进化的一个缩影,也为我们理解物种间免疫反应差异提供了新视角。
IFNAR2-S的出现无疑是基因组进化的一次神来之笔,它不仅丰富了灵长类动物的免疫调控能力,也提醒我们:基因组中的“小插曲”,常常孕育着伟大的生物学创新。
参考文献
Pasquesi GIM, Allen H, Ivancevic A, Barbachano-Guerrero A, Joyner O, Guo K, Simpson DM, Gapin K, Horton I, Nguyen LL, Yang Q, Warren CJ, Florea LD, Bitler BG, Santiago ML, Sawyer SL, Chuong EB. Regulation of human interferon signaling by transposon exonization. Cell. 2024 Dec 10:S0092-8674(24)01333-3. doi: 10.1016/j.cell.2024.11.016. Epub ahead of print. PMID: 39672162.