痤疮是发生于毛囊皮脂腺单位(pilosebaceous units,PSUs)的一种慢性炎症性疾病,是皮肤科最常见的疾病之一,影响约85%的青少年和成人,好发于面部、胸背部等皮脂溢出部位,以粉刺、红色丘疹、脓疱等为主要表现,严重者可伴有结节、囊肿,甚至留下永久性瘢痕。
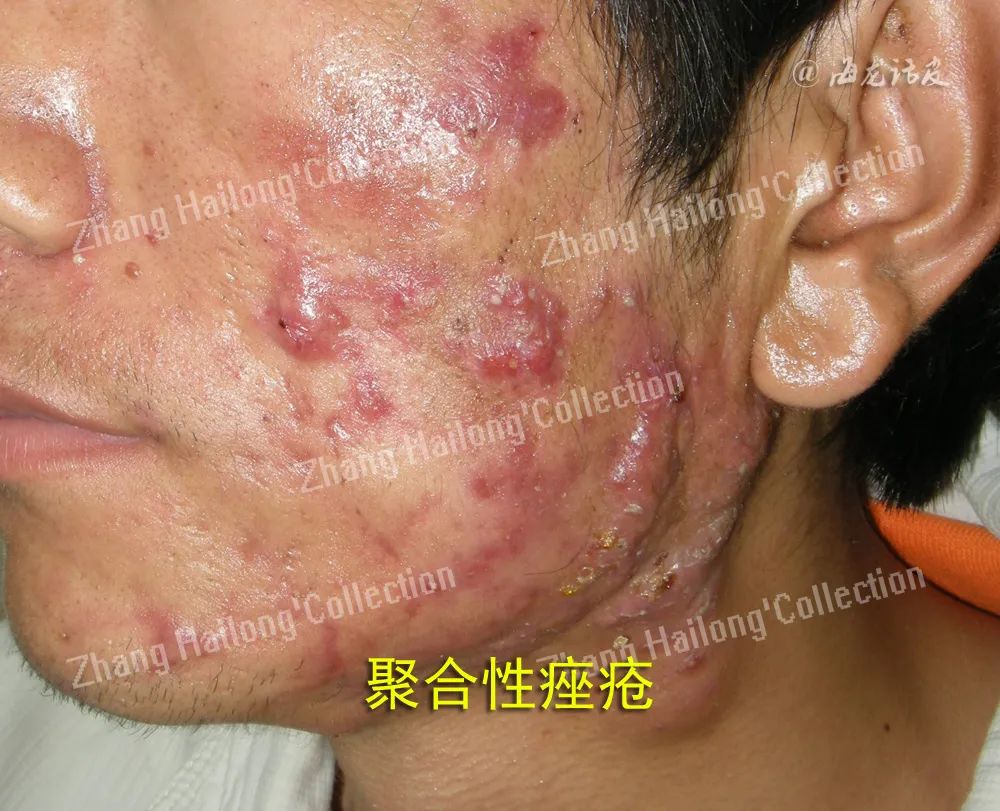
痤疮的发病机制复杂,尚未完全阐明,目前认为与皮脂分泌增加及皮脂溢出异常、毛囊皮脂腺导管异常角化、痤疮丙酸杆菌(Cutibacterium acnes,C.acnes)及皮肤微生物群失调、炎症与免疫等因素相关,此外雄激素、代谢以及环境、饮食、遗传等因素也可能引起痤疮的形成。炎症在痤疮的发病机制中起着核心作用,在炎症过程中,先天性免疫系统和适应性免疫系统协同激活以诱导免疫反应。
皮脂分泌及脂质代谢与炎症
痤疮的发展与皮脂分泌增加、皮脂溢出异常密不可分。雄激素能够刺激脂质合成以及皮脂腺细胞的增殖和分化,在痤疮发病中发挥作用。磷酸肌醇酸激酶(phosphatidyl inositol 3-kinase,PI3K)/Akt/叉头状转录因子O1(forkhead box O1,FoxO1)/雷帕霉素靶蛋白复合物1(mam-malian target of rapamycin complex1,mTORC1)通路可以维持皮肤稳态,调节细胞生长、增殖分化、凋亡等多种生理过程,该通路与各种免疫介导的炎症和过度增殖性皮肤病相关。
在痤疮中,雄激素、胰岛素样生长因子1(insulin growth factor 1,IGF-1)等可通过PI3K/Akt激活哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号转导。雄激素与位于细胞核内的雄激素受体(androgen receptor,AR)结合后,mTOR的磷酸化水平升高,并通过形成mTORC1催化核心,激活固醇调节元件结合蛋白-1(sterol regulatory element binding protein-1,SREBP-1),刺激脂质生成,其中mTOR是PI3K/Akt/FoxO1/mTORC1通路的关键下游元件。雄激素还负性调节内源性Wnt/β-catenin信号通路,Wnt/β-catenin靶基因c-MYC的表达上调,增强皮脂腺细胞分化。
分化中的皮脂腺细胞表现出高水平AR和过氧化物酶体增殖物激活受体(peroxisome proliferator activated receptors,PPARs),促进皮脂分泌增加,最终以全分泌方式将其内容物释放到皮脂腺导管中。胰岛素样生长因子1(insulin growth factor 1,IGF-1)信号在痤疮发病中起重要作用。IGF-1诱导的PI3K/Akt/FoxO1/mTORC1通路是痤疮发病机制中最重要的传导信号之一。
IGF-1通过PI3K/Akt/FoxO1/mTORC1途径和丝裂原活化蛋白激酶 (mitogen activated protein kinase,MAPK)增强PPARγ和SREBP-1的表达来诱导脂质合成,这些脂质代谢变化有利于C.acnes增殖及促进炎症的发生。以高糖碳水化合物和蛋白质摄入为特征的饮食也会增强IGF-1/PI3K/Akt/mTORC1信号表达。现在有研究认为IGF-1与雄激素在Akt激酶及mTORC1的激活中存在复杂的协同关系,共同导致Akt的激活。
FoxO1是痤疮发病机制中的另一个核心要素,胰岛素和IGF-1刺激PI3K/Akt级联反应,使得FoxO1从细胞核向细胞质转移,其不仅拮抗SREBP-1c的表达,而且通过抑制AR的反式激活来影响脂质的合成和皮脂腺的分泌。此外,FoxO1还可诱导腺苷5'-磷酸活化蛋白激酶(adenosine 5'-monophosphate-activated protein kinase,AMPK)通路的激活,是mTORC1的关键负调节因子;胰岛素和IGF-1通过调节FoxO1活性来增加脂质合成,最终参与痤疮发病。
同时IGF-1通过激活核转录因子-κB(nuclear transcription factor-κB,NF-κB)通路诱导皮脂腺细胞中的炎性细胞因子表达。除此之外,核苷酸结合寡聚化结构域样蛋白-1(nucleotide-binding oligomerization domain protein-1,NOD-1)在皮脂腺细胞中表达,其激动剂诱导IL-8表达,进一步激活NF-κB和MAPK信号通路诱导的炎症信号通路,具体机制有待进一步研究。
有关痤疮代谢组学研究结果表明,ATP结合盒式转运蛋白(ATP-binding cassette transporter,ABC)通路可能参与中度至重度痤疮的发病。ABC转运蛋白是依赖能量的跨膜转运蛋白,可通过打开和关闭转运循环来控制脂质颗粒的运输,以参与脂质的动态平衡,且ABC的表达会抑制SREBP,从而抑制脂肪形成。
此外,ABC转运蛋白在皮脂腺细胞中高度表达,其作用是合成和分泌皮脂,因此,ABC转运蛋白功能障碍可能通过调节皮脂的代谢和转运而参与痤疮的发病。痤疮的发病机制复杂,ABC转运蛋白如何在痤疮发病中发挥作用具体还需进一步研究明确。PI3K/Akt/FoxO1/mTORC1信号通路是皮肤炎症性疾病的新兴治疗靶点,但因其不良反应,迄今为止很少有药物通过测试并被批准用于治疗,因此需要进一步深入了解其病理机制,探索针对免疫介导炎症性皮肤病的安全且有效的治疗药物的研发。
毛囊皮脂腺导管角化异常及微粉刺的形成
在痤疮中,角质形成细胞的主要病理改变是过度增殖、异常脱屑和炎症介质的产生。有研究提出角化异常可能是由角质形成细胞凋亡不足引起。分泌过多的皮脂和脱落的角质形成细胞结合在一起逐渐形成角栓,并阻塞毛囊皮脂腺导管,最终形成微粉刺。微粉刺是所有痤疮的早期病理阶段,早期发生时在临床几乎不可见,仅可以在组织学水平看到。
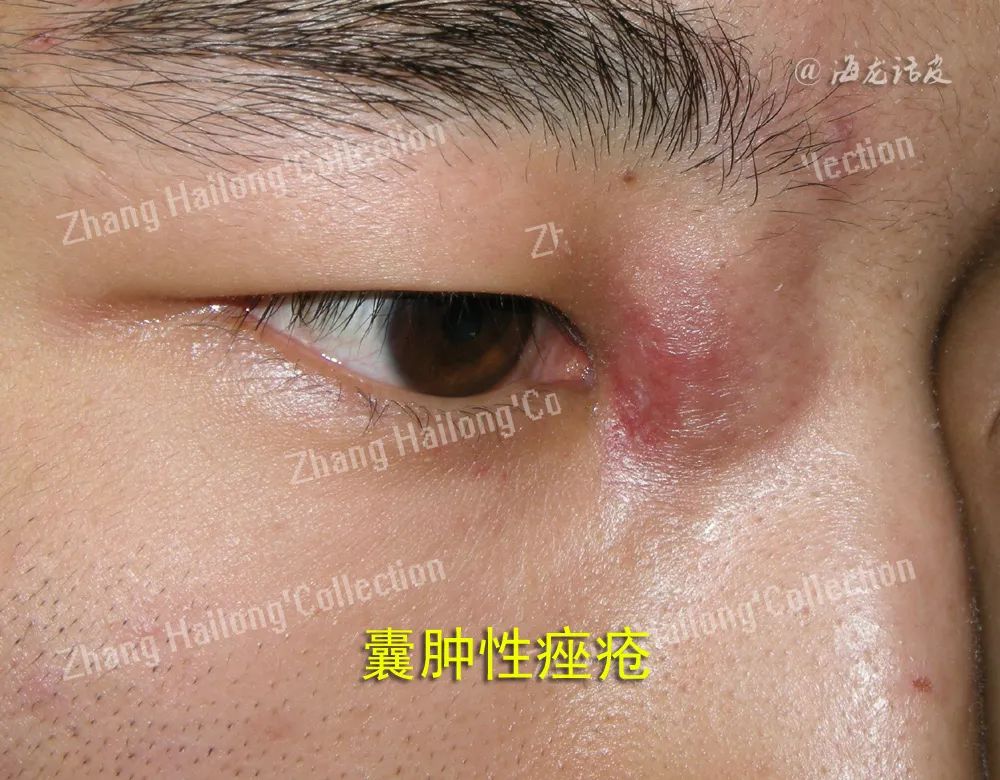
其形成可能与毛囊漏斗部角化异常、皮脂溢出增加及代谢异常相关。随着疾病的进展,微粉刺会演变成闭合性或开放性粉刺、炎性病变,最终可能形成结节或囊肿。微粉刺的数量和大小与痤疮的严重程度相关,粉刺病变中含有更多的微生物,这可能会导致更加显著的炎症。
有学者认为粉刺的形成是由于毛囊皮脂腺单位中角质形成细胞的积累和导管角质形成细胞的过度增殖所致。与皮脂腺细胞类似,角质形成细胞可能通过分泌炎症因子来参与炎症及先天免疫反应。C.acnes激活角质形成细胞上的Toll样受体(Toll like receptors,TLR)-2和TLR-4,进一步激活包括NF-κB通路和MAPK通路在内的信号级联反应。
随后,角质形成细胞产生白细胞介素(interleukin,IL)-1、IL-8、IL-6、粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony stimulating factor,GM-CSF)、肿瘤坏死因子α(tumor necrosis factor,TNF-α)、基质金属蛋白酶(matrix metalloproteinases,MMPs)和人β-防御素-2(human β-defensin-2,hBD-2)。除了TLR-2和TLR-4之外,角质形成细胞上表达的清道夫受体CD36也参与痤疮丙酸杆菌的识别。
痤疮丙酸杆菌被CD36识别后,角质形成细胞会迅速产生活性氧(reactive oxygen species,ROS),尤其是源自胞质酶NADPH氧化酶的超氧阴离子,超氧阴离子与一氧化氮结合形成过氧化亚硝酸盐,参与角质形成细胞裂解,这与IL-8的产生和角质形成细胞的凋亡相关,IL-8是痤疮发病中的一种重要促炎细胞因子,可见超氧阴离子在痤疮的炎症病变的 发展中起到重要作用。
看不见的微粉刺有一部分会发展为可见的皮肤病变,因此,微粉刺是预防痤疮发生的重要目标。现阶段,微粉刺的检测手段包括反射共聚焦显微镜、光学相关断层扫描等可用于临床。根据微粉刺的病因学,未来潜在的治疗方法可能涉及局部抗雄激素、IGF-1抑制剂等方法。
C.acnes与免疫炎症反应
C.acnes原本命名为痤疮丙酸杆菌(Propionibacterium acnes),是痤疮患者和正常人的主要皮肤共生细菌,在启动和维持痤疮炎症反应中至关重要,但学者对其引起炎症的机制尚有不同的观点。过去,C.acnes的定植被认为是皮脂腺细胞、角质形成细胞和单核细胞免疫反应的触发因素。
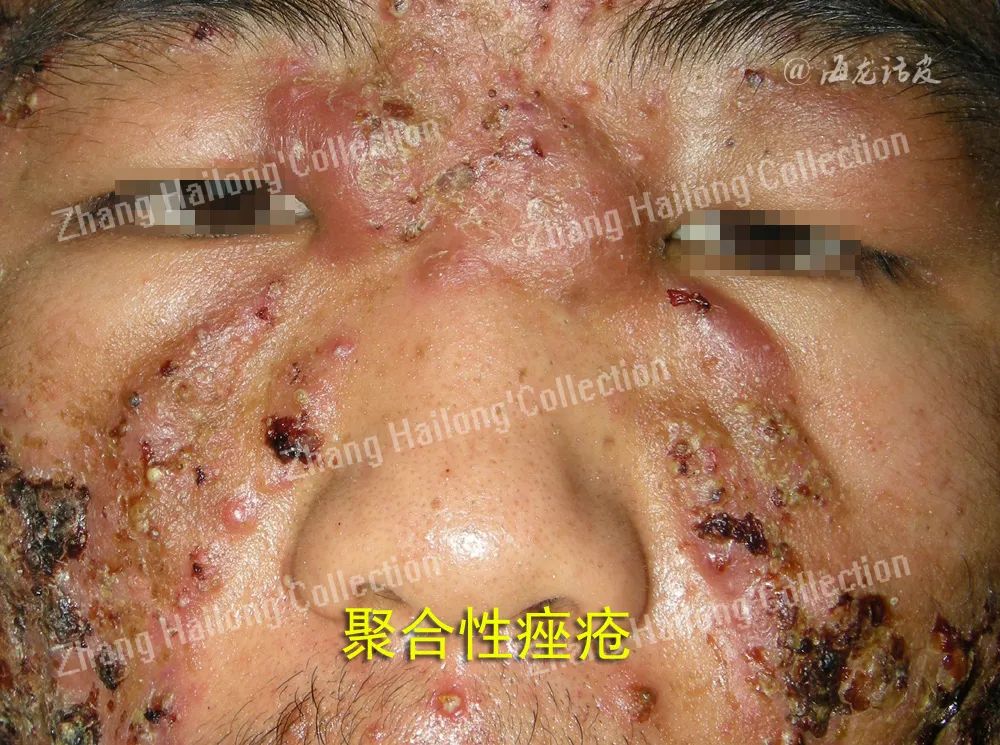
现在更多研究认为,痤疮的发病与C.acnes系统发育不平衡有关,皮肤微生物多样性的丧失以及先天免疫的激活可能会导致痤疮的慢性炎症,细菌释放的趋化物质将多形核白细胞吸引到炎症部位。这些细胞被局部激活,产生 炎性细胞因子,如TNF-α、IL-1β和IL-8。痤疮发病机制的复杂性还在于先天性免疫及适应性免 疫的相互作用。
C.acnes通过单核细胞/巨噬细胞谱系细胞上表达的TLR-2进行识别,TLR-2诱导IL-20p40启动子活化并产生IL-12和IL-8。TLR-2通过其细胞外结构域识别C.acnes的相关分子结构(例如肽聚糖和脂蛋白)来启动信号传导。随后,侵入信号从胞外结构域的二聚体转运至胞内Toll/IL-1受体结构域,后者会募集适配蛋白,包括髓样分化因子88(myeloid differentiation factor 88,MyD88)和MyD88的衔接子蛋白(MyD88-adapter-like protein,Mal)。
MyD88导致IL-1受体相关激酶磷酸化,随后促进肿瘤坏死因子受体相关因子6(TNF receptor associated factor 6,TRAF6)活化,TRAF6通过募集转化生长因子活化激酶1(transforming growth factor-activated kinase 1,TAK1)和TAK1结合蛋白2(TAK1-binding protein2,TAB2)促进经典NF-κB通路的活化,诱导产生IL-1β、IL-6、IL-8、TNF-α等炎症因子。
C.acnes还上调促炎症蛋白酶caspase-1和核苷酸结合寡聚化结构域样受体家族pyrin结构域3(NOD-like receptor family pyrin domain-containing 3,NLPR3)的表达,同时诱导单核细胞-巨噬细胞NLRP3炎症小体的激活,虽然NLRP3炎症小体激活的具体机制尚未完全阐明,但目前已明确的是与活性氧(reactive oxygen species,ROS)的产生和钾的流出相关,最终大量产生IL-1β,以此触发先天免疫反应。
C.acnes诱导的ROS可引发NF-κB和MAPK通路的活化,进而诱导巨噬细胞中诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)/一氧化氮(nitric oxide,NO)和环氧合酶-2(cyclooxygenase-2,COX-2)/前列腺素E2(prostaglandin E2,PGE2)的信号的表达。在TLR-2和C.acnes之间的相互作用中,NF-κB是一种重要的TLR-2下游信号,其负责与痤疮和先天免疫应答相关的许多炎症基因,通过释放各种促炎细胞因子,如COX-2、iNOS、IL-1β 和TNF-α等,这些细胞因子在痤疮的毛囊过度角化和炎症反应中起主要作用,对痤疮的炎症反应产生重大影响。
C.acnes与痤疮发生初期Th17免疫炎症途径的激活存在密切联系,参与C.acnes诱导的适应性免疫反应的淋巴细胞是CD4+T细胞,其在C.acnes诱导下分化为Th17细胞,Th17细胞是IL-17释放的起源。同时C.acnes触发外周血单核细胞分泌IL-1β、IL-6和转化生长因子-β(transforming growth factor-β,TGF-β),这些炎性介质也诱导幼稚CD4+CD45RAT细胞分化为Th1及Th17细胞。被诱导释放IL-17和干扰素IFN-γ(Th效应细胞因子),促进Th17和Th1/Th17宿主联合反应,从而导致痤疮的炎症反应。
除了炎症介质外,中性粒细胞还可以通过增加过氧化氢的产生来介导炎症。得益于这些发现,可以考虑更有针对性、更有效且副作用更小的治疗方式,从而提高痤疮患者的治疗依从性和生活质量。比如补充益生菌、微生物组修饰等疗法恢复皮肤微生物群多样性,直接改变皮肤微生物群和炎症免疫反应; 开发痤疮靶向治疗,识别疾病发病机制的生物标志物等。除了针对痤疮发生机制的治疗外,实施皮肤护理以避免因皮肤刺激而出现新的炎症病变,对于治疗的成功实现也至关重要。
结语与展望
痤疮是一种慢性炎症性皮肤病,其发病机制复杂,多种因素交错。近年来的研究提高了我们对微生物、炎症与免疫等与痤疮发病关系的理解,但我们对痤疮发展的各个阶段炎症及免疫活动的理解仍然有限。中重度痤疮可造成患者身体、心理等方面的影响,抗生素及维A酸类药物长期以来一直是痤疮的主要治疗方法,这些治疗方法一方面对痤疮的治疗效果有限,另一方面治疗的不良反应,如维A酸类药物对生殖的影响、抗生素耐药性产生及可能引起皮肤生态失调等,都对痤疮的治疗有所限制。
对痤疮的发病机制以及参与 其中的信号通路的研究为痤疮治疗的新靶点提供 了思路,未来的治疗可能不再旨在“杀死”痤疮丙酸杆菌,而是维持皮肤微生物群的平衡,控制早期炎症及免疫反应,不再让痤疮发展至囊肿、瘢痕等损容性阶段。
参考文献:
1.刘白,吴瑾.痤疮发病相关信号通路的研究进展[J].皮肤性病诊疗学杂志, 2024,11(31):795-800.
2.中国痤疮治疗指南专家组.中国痤疮治疗指南(2019修订版)[J].临床皮肤科杂志,2019,48(9) : 583-588.
3.郎德休,廖勇,杨蓉娅.痤疮丙酸杆菌的新命名及其与寻常痤疮的相关性[J].中华皮肤科杂志,2020,53(5) : 394-397.
4.其他文献略.