摘要:在过去的40年中,大量的研究显示,细胞Ca2 +稳态的丧失在缺血性脑损伤的发病机制中扮演重要作用。细胞内Ca2 +超载可导致活性氧的生成增加、调节自噬、激活钙蛋白酶等。线粒体Ca2 +摄取被证明可以缓冲细胞内Ca2 +水平,随之而来的线粒体Ca2 +超载可调节线粒体生物学效应从而进一步调控细胞存活。作者结合缺血性脑损伤中神经元Ca2 +稳态丧失特点,详细分析了Ca2 +转运途径及各转运体的治疗研究进展,为缺血性脑损伤的研究和分子干预提供新的思路。
卒中是继缺血性心脏病之后的第二大死亡原因,也是致残的主要原因,其中缺血性卒中是卒中各型中发病率最高的一种。根据脑组织缺血的严重程度分为缺血核心区和缺血半暗带区,其中缺血核心区向缺血半暗带区扩展可以通过药物来预防。根据既往研究,有超过1 000 种神经保护剂在卒中模型中具有很好的神经保护作用,但仅114 种转化到临床,其中只有阿替普酶被证明可以改善患者的神经功能预后。有研究者提出,在缺血性脑损伤时,神经保护以及神经损伤的修复为多种机制联合。细胞Ca2 +稳态丧失是缺血性脑损伤的病理生理机制之一,其犹如一条线,串联缺血性脑损伤的各个病理生理机制,如细胞酸中毒、线粒体功能障碍、氧化应激、自噬、凋亡及激活钙蛋白酶等。因此,深入研究细胞Ca2 +稳态丧失可能为多机制联合治疗提供思路。然而在缺血性脑损伤的不同阶段,细胞内Ca2 +(intracellular Ca2 +,[Ca2 +]i)水平变化不尽相同,其中涉及复杂的Ca2 +摄取与输出途径,因此笔者首先介绍了缺血性脑损伤时[Ca2 +]i及线粒体内Ca2 +(matrix Ca2 +,[Ca2 +]mt)浓度的变化,分析其可能涉及的Ca2 +转运途径及病理生理作用,并基于Ca2 +转运途径重点阐述了Ca2 +转运体相关治疗研究进展,最后对[Ca2 +]i 稳态失衡在缺血性脑损伤中的研究进行了展望。
1 缺血性脑损伤时[Ca2 +]i水平的变化
1. 1 脑缺血期[Ca2 +]i水平的变化
在脑缺血期,神经元[Ca2 +]i将经历2 次上升。通过离子选择性微电极评价完整大鼠脑组织神经元[Ca2 +]i水平变化,结果显示[Ca2 +]i水平在脑缺血后并未立即上升,而是在细胞内pH 值出现碱性位移后,[Ca2 +]i 水平才出现初次小幅度上升,但此时,细胞外Ca2 +(extracellular Ca2 +,[Ca2 +]e)浓度还未开始下降。在[Ca2 +]i 小幅度的上升之后,会经历一段“安静”阶段,随后[Ca2 +]i开始第2 次大幅度上升。由于[Ca2 +]i浓度上升早于[Ca2 +]e浓度下降,因此可能存在除[Ca2 +]e内流之外的其他途径导致[Ca2 +]i 浓度升高,比如细胞内储存Ca2 +的释放。“安静”阶段之后的[Ca2 +]i 水平升高,应该与[Ca2 +]e内流密切相关。
1. 2 再灌注期[Ca2 +]i水平的变化
再灌注损伤期[Ca2 +]i水平是否恢复至缺血前水平与脑电图恢复情况和脑组织病理学改变有关。有研究表明,再灌注之后,大鼠海马CA1 区[Ca2 +]i浓度在10 min内下降至缺血前水平。但也有研究报道,在大脑中动脉闭塞1 h后再灌注30 min时,雄性猫皮质中[Ca2 +]i 浓度变化与缺血性脑损伤的组织病理学变化相关。在该实验中,根据再灌注前30 min脑电图振幅恢复情况将实验动物分为2个亚型:1 型动物恢复10%及以上,2 型动物恢复小于10%。再灌注时,1 型动物的[Ca2 +]i 信号显著下降,而2型动物的[Ca2 +]i信号未下降,反而进一步增加。通过半定量分级系统评估脑组织缺血性改变的程度也发现,2 型亚组的组织病理学改变程度明显大于1 型亚组。在光学显微镜下具体表现为:1型亚组中,5 只动物均为脑皮质轻微受累(出现少量散在的缺血性神经元),然而在2 型亚组中,有1只为脑皮质严重受累(含有大量的缺血性神经元),3只为脑皮质出现中度受累(含有数个缺血性神经元),仅有1 只为脑皮质轻微受累。这可能与再灌注后1 型亚组质膜上Ca2 +输出途径迅速恢复有关,而2型亚组则没有,导致2 型亚组[Ca2 +]i进一步积累。
1. 3 [Ca2 +]mt水平的变化
N-甲基-D-天冬氨酸(N-methyl-D-aspartate,NMDA)可诱导[Ca2 +]mt水平升高。在一项体外实验中,用低浓度(20 μmol / L)或高浓度(200 μmol / L)的NMDA刺激SD胎鼠海马神经元60 s,检测海马神经元单细胞中[Ca2 +]i水平发现,结果显示,低浓度和高浓度的NMDA均可使[Ca2 +]i水平增加,增加的峰值分别为(518 ±47)nmol/ L和(1 396 ±392)nmol/ L。检测海马神经元细胞群中[Ca2 +]mt,结果显示,低浓度和高浓度的NMDA均可使[Ca2 +]mt增加,其中高浓度NMDA使其增加的程度更为显著[200 μmol / L诱导的[Ca2 +]mt 改变/ 20 μmol / L 诱导的[Ca2 +]mt改变=(1. 7 ± 0. 3),P < 0. 05)]。这些结果提示,[Ca2 +]i水平和[Ca2 +]mt 的变化程度均与NMDA浓度有关。
2 缺血性脑损伤时Ca2 +的摄取和输出途径
2. 1 神经元对Ca2 +的摄取途径
脑缺血时,[Ca2 +]e转移至细胞内,[Ca2 +]i浓度急剧上升。[Ca2 +]e转移至细胞内是由不同的受体和通道介导的,包括酸敏感离子通道(acid sensing ion channels, ASICs)、谷氨酸受体(Glutamate receptor,GluR)、电压门控钙通道(voltage-gated calcium channels, VGCCs)、 Na + -Ca2 + 交换体(Na + -Ca2 +exchanger,NCX)、瞬时受体电位(transient receptor potential,TRP)通道等。
2. 1. 1 ASICs:ASICs是Na +选择性阳离子通道,在大脑皮质、小脑、海马、杏仁核和嗅球中广泛表达,目前已鉴定出6种ASICs亚基蛋白,其中ASIC1a可根据细胞外pH变化,调节快速瞬态内向电流。ASIC1a被激活的pH阈值为7. 0,随着pH降低,ASIC1a可进一步被激活,激活后的ASIC1a可介导[Ca2 +]e内流。脑缺血时,无氧葡萄糖代谢增强和腺苷三磷酸(adenosine triphosphate,ATP )水解释放H +导致乳酸积累,由此产生的酸中毒导致缺血核心区细胞外的pH 值从7. 3 下降至6. 0 ~ 6. 5,导致ASICs激活,诱导Na +和[Ca2 +]e内流。ASIC1a激活介导的细胞外Na +和[Ca2 +]e内流甚至有可能早于GluR激活介导的细胞外Na +和[Ca2 +]e 内流。因为在缺血导致细胞外pH值低于7. 0时,[Ca2 +]e水平还未开始明显下降,而此时ASIC1a已被激活。
2. 1. 2 GluR:GluR 主要包括α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体(α-amino-3-hydroxy-5-methyl-4-isoazole propionic acid receptor,AMPAR)、N-甲基-D-天冬氨酸受体(N-methyl-D-aspartate receptor,NMDAR)、红藻氨酸受体和代谢型受体。正常情况下,Mg2 +会阻断NMDAR。但是在AMPAR 激活后,Na +内流,造成膜内Na +浓度急剧增加,引起突触后膜去极化,解除了Mg2 +对NMDA 门控通道的阻断作用,导致[Ca2 +]e 通过NMDA 门控通道内流。AMPAR除了参与促进[Ca2 +]e通过NMDA门控通道流入的过程,本身也介导[Ca2 +]e内流。AMPAR是由GluR1 ~ 4编码的亚基组成的异聚体,其中GluR2可阻止[Ca2 +]e通过AMPAR。脑缺血时,GluR2 mRNA在脆弱神经元中表达下调,导致Ca2 + 渗透性AMPAR 形成,介导[Ca2 + ]e内流。
2. 1. 3 VGCCs及NCX:随着ASICs、GluR 激活,细胞外Na +和[Ca2 +]e大量内流,细胞膜去极化,将触发雪崩式的反应,激活一系列电压依赖性内流途径,比如[Ca2 +]e通过L和T型VGCC进入。NCX具有双向转运功能,以Ca2 +外排(正向)或Ca2 +内流(反向)方式运输Na +和Ca2 +,其转运方向依赖于跨膜离子浓度以及膜电位。在神经元缺血时,Na +和Ca2 +离子干扰有利于NCX反向工作,导致[Ca2 +]i水平增加;但是在再灌注时NCX可能恢复正向模式的活性,参与[Ca2 +]i外排。
2. 1. 4 TRP 通道家族:TRP 是非特异性阳离子通道,构成一大类质膜Ca2 +通道,可分为6个亚家族,分别为经典家族(canonical,TRPC)、香草醛类受体家族(vanilloid,TRPV)、前列腺特异性基因黑素抑素家族(melastatin,TRPM)、多囊蛋白家族(polycystin,TRPP)、黏脂蛋白家族(mucolipin,TRPML)和锚蛋白家族(ankyrin,TRPA)。TRP通道的激活允许[Ca2 +]e进入细胞,导致细胞去极化,从而可能参与了VGCCs 的激活[22]。神经元损伤期间,NMDAR的激活导致[Ca2 +]e 进入,刺激自由基的生成。而TRP 通道,如TRPM2、TRPV 和TRPC等可被过氧化氢等氧化应激产物激活。随着TRP通道的开放,[Ca2 +]e 内流,[Ca2 +]i 水平升高,可刺激线粒体对Ca2 +的摄取,进一步加重氧化应激。此外有研究显示,TRPV 家族还可通过其他途径参与[Ca2 +]e内流。如在小鼠大脑中动脉栓塞模型中发现,TRPV4 可通过增强海马神经元中NMDAR 功能,参与谷氨酸介导的兴奋性毒性,促进神经元对Ca2 +的渗透性;TRPV3 也参与[Ca2 +]e内流,其半最大激活pH值为6. 1。
脑缺血时,[Ca2 +]i 浓度的上升除与[Ca2 +]e内流有关外,还可能与细胞内储存Ca2 +的释放有关。[Ca2 +]e 进入神经元内,可由肌质网/内质网Ca2 + -ATP酶泵入内质网进行储存。大脑中动脉栓塞模型大鼠在缺血后的前4 h内,细胞外腺苷和ATP的浓度增加,其浓度能够刺激P2 受体。P2 受体分为P2X离子型和P2Y代谢型受体。P2X受体存在于细胞表面,对Ca2 +具有非选择性渗透性;P2Y受体与Gq / 11蛋白偶联,可激活磷脂酶C,并促进磷酸肌醇级联反应,与之伴随的是细胞内三磷酸肌醇储存和内质网Ca2 +释放。同时内质网的兰尼碱受体兴奋也是细胞内储存Ca2 +的释放通道之一。在再灌注损伤期,PC12 细胞[Ca2 +]i浓度随着肌质网/内质网Ca2 + -ATP 酶表达的下调而升高。因此肌质网/内质网Ca2 + -ATP酶也可能参与了缺血性脑损伤中神经元Ca2 +稳态失调。结合本文所述,总结如图1。
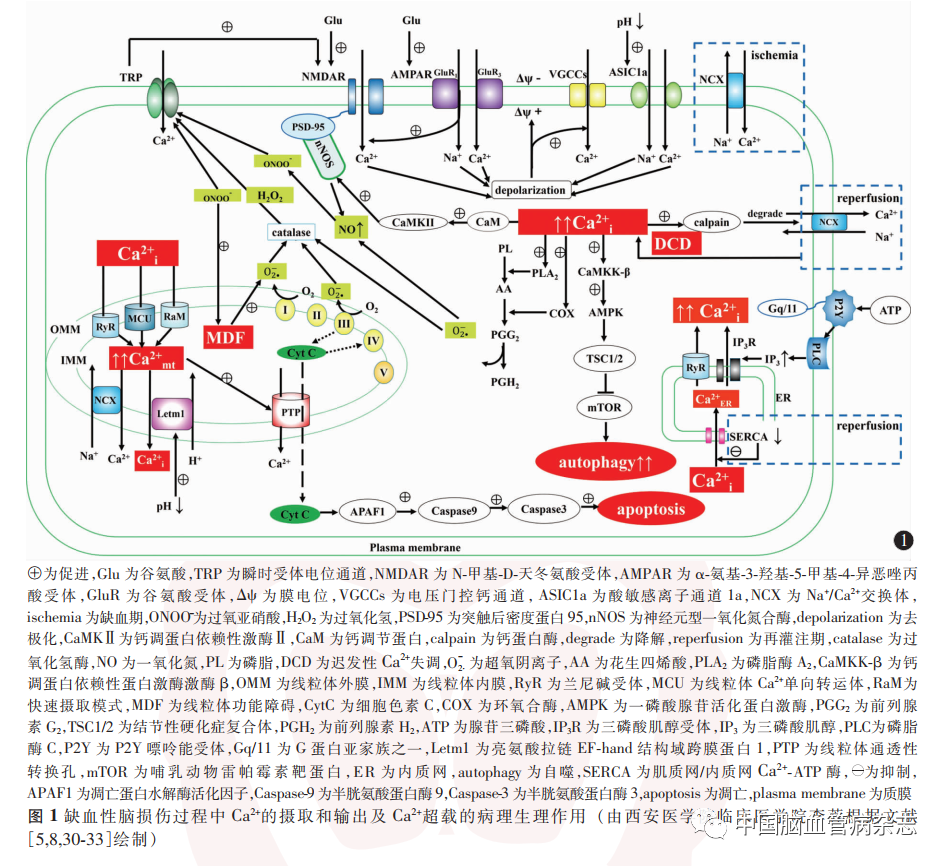
2. 2 星形胶质细胞对神经元Ca2 +超载的作用
星形胶质细胞最初被认为不参与神经元活动的调节,然而越来越多的研究表明,星形胶质细胞可通过增加自身[Ca2 +]i 水平响应神经元活动,调节神经元[Ca2 +]i 水平。既往研究显示,通过模拟体内缺氧、缺糖条件,可导致星形胶质细胞[Ca2 +]i水平升高[37]。在复氧阶段,海马星形胶质细胞中Na + / H +交换体异构体1的活化可以与NCX的反向模式偶联,导致海马星形胶质细胞[Ca2 +]i 水平显著增加[在复氧3 h时从(44. 2 ± 12. 1)nmol / L增加至(416. 7 ± 43. 9)nmol / L,并在复氧5 h 时继续增加,P < 0. 05],同时Na + / H +交换体异构体1的活化导致细胞内Na +水平升高,进而降低谷氨酸转运体对谷氨酸的摄取,甚至通过谷氨酸转运体的反向作用导致谷氨酸释放。由于星形胶质细胞导致谷氨酸摄取受损或释放增加,可引发NMDAR和AMPAR的过度刺激,导致神经元[Ca2 +]i稳态丧失。
在星形胶质细胞中表达的TRP 通道家族也参与了神经元Ca2 +稳态失衡的进展。O2 可诱导促进星形胶质细胞质膜上TRPA1 蛋白的内化。在缺氧条件下,TRPA1 蛋白内化受阻并在质膜上蓄积,促进星形胶质细胞中含ATP 囊泡区室的胞吐作用加强,驱动ATP 的释放,导致细胞外ATP 的浓度增加。如前所述,细胞外ATP浓度的增加可进一步通过磷酸肌醇级联反应参与神经元Ca2 +稳态调节。
2. 3 线粒体对Ca2 +的摄取和输出途径
线粒体对[Ca2 +]i 浓度的调节是可逆的,当[Ca2 +]i浓度高于“设定点”时,线粒体起到储存的作用,当[Ca2 +]i浓度低于“设定点”时,线粒体释放Ca2 +,在此调节过程中主要涉及的是线粒体对Ca2 +的摄取与输出。Ca2 +积累到线粒体基质需跨越两层膜,即线粒体外膜和线粒体内膜。由于线粒体外膜上的电压依赖性阴离子选择通道对Ca2 +是可渗透的,因此线粒体对Ca2 +的摄取问题主要集中在线粒体内膜的运输系统上。
2. 3. 1 线粒体Ca2 +摄取:线粒体对Ca2 +摄取主要通过线粒体Ca2 + 单向转运体(mitochondrial Ca2 +uniporter,MCU)实现。MCU 位于线粒体内膜,是一种门控离子通道,对Ca2 +有较高的选择性,通过内向整流,使Ca2 +进入线粒体。MCU对Ca2 +摄取由线粒体膜电位驱动,活性随着线粒体外游离Ca2 +浓度的升高而迅速增加。线粒体还可以通过“快速摄取模式”摄取Ca2 +。与MCU相比,“快速摄取模式”的特点为持续时间很短的快速吸收,因此,MCU相对来说是一种“较慢摄取模式”。由于“快速摄取模式”持续时间很短,在摄取之后迅速被抑制,因此在不到1 s后,唯一观察到的Ca2 +摄取通道是MCU。在离体心脏线粒体中,兰尼碱可抑制线粒体对Ca2 +的摄取,抑制Ca2 +超载所引起的线粒体肿胀,并发现线粒体的兰尼碱受体与线粒体Ca2 +转运蛋白结构相似,因此除了上述的“快速摄取模式”和“较慢摄取模式”,兰尼碱受体可能参与了线粒体Ca2 +内流。见图1。
2. 3. 2 线粒体Ca2 +输出:关于线粒体中Ca2 +从基质中的输出途经,目前存在多种机制。一项体外研究表明,添加Na +可激活Ca2 +从脑线粒体流出,并且线粒体Na + / Ca2 +交换体抑制剂CGP37157 可阻断脑线粒体中Ca2 +流出,因此神经元线粒体中Ca2 +的释放主要为Na +依赖性,受线粒体Na + / Ca2 +交换体调节。研究表明,长期或反复暴露于高水平的Ca2 +中,是线粒体通透性转换孔(permeability transition pore,PTP)开放和线粒体去极化的基础。PTP打开后允许小分子量代谢物、辅因子和离子在线粒体基质和胞质溶胶之间达到平衡,这其中就包括Ca2 + 。2009年,通过全基因组果蝇RNA干扰筛选实验鉴定出亮氨酸拉链EF-hand结构域跨膜蛋白1 为一种假定的Ca2 + / H + 反向转运体,介导[Ca2 +]mt转运。亮氨酸拉链EF-hand 结构域跨膜蛋白1相关的[Ca2 +]mt调节受pH梯度的限制,线粒体外部的酸性环境可驱动[Ca2 +]mt的释放,而碱性环境可促进线粒体对Ca2 +的摄取。见图1。
3 Ca2 +超载的病理生理学作用
3. 1 线粒体功能障碍
[Ca2 +]i 超载与线粒体功能障碍密切相关。[Ca2 +]mt积累可以触发线粒体通透性转变,PTP打开。尤其在缺血-再灌注的情况下,细胞中的大多数线粒体同时经历PTP 打开。持续而广泛的PTP开放促使分子量小于1 500 g / mol 的溶质扩散至细胞质,引发H2O的反向分布。H2O进入线粒体基质导致线粒体嵴部分膨胀和基质体积扩大,导致线粒体酶和代谢物稀释。随着质子流入基质,线粒体膜电位耗散,进而促使三羧酸循环和呼吸复合物功能被抑制,影响ATP的合成。此外,永久性PTP开放可导致线粒体外出现还原型烟酰胺腺嘌呤二核苷酸(磷酸)并抑制呼吸链。而且PTP的开放可促进活性氧类(reactive oxygen species,ROS)的产生,且ROS的产生可能促进线粒体电子呼吸链超级复合物的解体,随着电子呼吸链的受损,特别是在呼吸复合物Ⅰ 内,线粒体ROS产生将进一步增加。ROS水平的持续增加又对线粒体是有害的,线粒体受损又进一步促进ROS的产生(这种现象即为“ROS 诱导ROS 产生”),最终引起更广泛的损害。
3. 2 凋亡
[Ca2 +]mt超载与细胞凋亡密切相关。在一项体外实验中,NMDA暴露后,[Ca2 +]mt浓度升高,原定位于线粒体的细胞色素C 可通PTP从线粒体中释放,而当减少[Ca2 +]mt积累时,可阻止线粒体细胞色素C的释放,提示[Ca2 +]mt超载可能与线粒体亚群中凋亡因子的释放有关。但是也有研究显示,细胞色素C的释放可能与PTP无直接关系。细胞色素C进入细胞质后,可与凋亡蛋白水解酶活化因子结合形成复合物,激活半胱氨酸蛋白酶9,活化的半胱氨酸蛋白酶9进一步激活半胱氨酸蛋白酶3,从而启动半胱氨酸蛋白酶家族引发的级联反应,导致凋亡发生。除此之外,线粒体肿胀引起的线粒体外膜完整性丧失,促使线粒体凋亡诱导因子1和核酸内切酶G等因子的释放,从而启动了细胞凋亡的内在途径,最终导致细胞死亡。见图1。
3. 3 氧化-亚硝化应激
通过NMDAR内流的Ca2 +可有效地刺激神经元型一氧化氮合酶(neuronal nitric oxide synthase,nNOS),导致nNOS激活,调节一氧化氮的产生。这可能与nNOS通过PDZ-PDZ与支架蛋白突触后密度蛋白95的PDZ2相互作用有关。脑缺血时,随着NMDAR 的激活,Ca2 + 内流并与钙调节蛋白(calmodulin,CaM)结合形成Ca2 + -CaM,从而激活钙调节蛋白依赖性激酶Ⅱ,开启nNOS 的磷酸化级联反应,促进一氧化氮的产生。而超氧阴离子很容易与一氧化氮反应,形成高度反应性的过氧亚硝酸,其氧化能力远远超过超氧阴离子和一氧化氮,过氧亚硝酸的一个重要靶点为线粒体,其导致线粒体功能障碍使氧自由基的生成进一步增多。其次,在缺血期,一些Ca2 +依赖的酶被激活,如磷脂酶A2 和环氧合酶的激活。磷脂酶A2 激活导致磷脂释放花生四烯酸,环氧化酶催化两个O2 分子加入花生四烯酸生成前列腺素G2,前列腺素G2 迅速过氧化为前列腺素H2,同时释放超氧阴离子。见图1。
3. 4 钙蛋白酶
[Ca2 +]i超载导致钙蛋白酶的激活,钙蛋白酶是一种Ca2 +依赖性蛋白酶,可降解神经元中至关重要的蛋白质,导致神经元死亡。在细胞中,钙蛋白酶具有不同的底物,包括细胞骨架元素、钙转运通道的组成部分(如NMDAR、谷氨酸/ AMPAR和质膜Ca2 + -ATP酶)。谷氨酸作用于小脑颗粒神经元可促进钙蛋白酶被激活,钙蛋白酶的激活又可促进NCX3的降解,进一步导致细胞质对[Ca2 +]i清除机制下降,而这与介导神经元死亡的迟发性Ca2 +失调密切相关。见图1。
3. 5 自噬
[Ca2 +]i超载在细胞自噬中具有重要的调节作用。有研究报道,Ca2 +可能是通过Ca2 + /钙调蛋白依赖性蛋白激酶激酶β /磷酸腺苷活化蛋白激酶(Ca2 + / calmodulin-dependent protein kinase kinase β /adenosine monophosphate-activated protein kinase,Ca2 + / CaMKK-β / AMPK)通路触发自噬,CaMKK-β是一种Ca2 +活化激酶,也是AMPK的直接激活剂。在低ATP条件下,CaMKK-β以AMPK和结节性硬化症复合体1 / 2 依赖性方式触发哺乳动物雷帕霉素靶蛋白的抑制,从而触发自噬。见图1。
4 Ca2 +转运体相关治疗研究进展
4. 1 ASICs
ASIC1a是哺乳动物大脑中的主要酸传感器,也是脑缺血后酸中毒诱导的神经元损伤的关键介质。ASIC1a 基因消融和选择性药理学抑制可减少啮齿动物缺血性卒中后的神经元死亡。早期研究显示,阿米洛利、布洛芬和氟比洛芬也是ASIC1a抑制剂,其可抑制酸介导的[Ca2 +]i增加,并减少梗死体积,改善神经功能缺损。迄今为止最有效的ASIC1a阻断剂为狼蛛毒素,脑室内给予单次小剂量狼蛛毒素,可减少大脑中动脉闭塞后72 h时约70%的皮质梗死体积并恢复至正常水平的神经和运动功能。但是狼蛛毒素和其他ASIC抑制剂均未显示卒中发生2 ~ 4 h 后给药具有显著的神经保护作用。2017 年的一项研究表明,Hi1a 在缺血性卒中的局灶模型中具有高度的神经保护作用,其通过一种独特的作用方式抑制ASIC1a,并在卒中发生后8 h内给药均具有保护作用。
4. 2 GluR
谷氨酸的释放增加和(或)摄取减少可导致神经元Ca2 +稳态失调。在缺血性损伤时,星形胶质细胞中双孔钾离子通道家族中的TREK-1 蛋白表达上调,抑制其活性可导致谷氨酸摄取受损,星形胶质细胞和神经元共培养物中的神经元凋亡增加。这些证据表明,TREK-1通过调节星形胶质细胞的功能参与缺血后的神经元结局,因此TREK-1 可作为治疗缺血性卒中的靶点。NMDAR 主要由NR1 和NR2A ~ D亚基组成,突触NR2A表达增加促进神经元的存活,而NR2B 表达增加促进神经元凋亡并增加对兴奋性毒性的敏感性,蝎毒耐热蛋白可通过抑制NR1和NR2B的表达以及防止NR2A表达减少而发挥抗神经毒性作用。黄体酮等类固醇类和内源性神经营养肽垂体腺苷酸环化酶激活多肽27 / 38也可通过调节NMDAR亚基表达而发挥神经保护作用。有研究显示,缺血性卒中大鼠脑中HECT结构域E3 泛素蛋白连接酶4 的下调可通过黏膜相关淋巴样组织淋巴瘤易位蛋白1 促进NR2B的磷酸化,导致Ca2 +超载。因此HECT 结构域E3泛素蛋白连接酶4 和黏膜相关淋巴样组织淋巴瘤易位蛋白1 可能是预防NR2B活化的潜在靶点。虽然NMDAR长期以来一直被认为是缺血性卒中的治疗靶点,但是NMDAR 拮抗剂在临床试验中的应用使卒中患者产生了严重的不良反应。而且目前研究者已经认识到,其还具有重要的生理功能,如促进神经元存活,因此完全阻断NMDAR可能是有害的。因此寻找可以抑制NMDAR过度活跃,又不影响其生理功能的抑制剂成为新的治疗靶点。有研究显示,α2δ-1 对于缺血诱导的神经元NMDAR过度活跃必不可少,而解偶联α2δ-1-NMDAR可以在减缓缺血-再灌注诱导的脑损伤的同时不影响NMDAR的生理活性。因此,α2δ-1 结合的NMDAR可作为治疗缺血性卒中的靶点。缺血-再灌注后质膜上含GluA2的AMPAR丢失可导致[Ca2 +]e内流,并与延迟性神经元死亡密切相关。2021 年的一项体外研究表明,氧化应激可能是缺血-再灌注诱导的AMPAR内化和降解的基础,其发现锰(Ⅲ)-四(4-N-甲基吡啶基)卟啉(一种超氧化物歧化酶模拟物)预处理可减弱体外氧葡萄糖剥夺-复氧诱导的GluA2 亚基的内化,减弱GluA2 亚基降解,因此氧化应激可能在缺血-再灌注损伤后AMPAR介导的细胞死亡中起重要作用,有待于进一步研究。
4. 3 VGCCs及NCX
随着GluR激活,细胞膜去极化,VGCCs被激活,进一步加重[Ca2 +]e内流。而普瑞巴林可与VGCC的α2δ 亚基特异性结合,从而减少谷氨酸释放及[Ca2 +]e内流。然而由于给药剂量、途径的不同,会对脑梗死体积和神经功能的评分产生不同的结果,因此需要进一步研究确定其作用效果。NCX1、NCX2和NCX3是NCX家族的3个不同基因。NCX的反向工作可导致[Ca2 +]i水平增加。棕榈酰化为目前唯一已知的可调节NCX活性的可逆翻译后修饰,非棕榈酰化NCX对失活的敏感性降低,最终导致[Ca2 +]i水平升高,因此,NCX的棕榈酰化状态可调节[Ca2 +]i水平。而胰岛素可促进锌指样DHHC结构域蛋白5活性位点的棕榈酰化,从而导致NCX1棕榈酰化增强,调节NCX1 失活,进一步调控[Ca2 +]i 水平。三碘甲状腺原氨酸预处理可抑制NCX反向工作,减少[Ca2 +]e内流,以维持Ca2 +稳态和保护心肌细胞免受缺血-再灌注损伤。既往研究表明,NCX抑制剂ORM-10103可通过抑制NCX的反向模式,防止毒毛旋花子苷元诱导的Ca2 +积累和自发心室舒张期Ca2 +释放。此外KB-R7943和SEA-0400在早期也被认为是“准选择性”NCX 阻滞剂,但这些主要应用在心律失常的研究中,因此上述抑制剂在脑缺血性损伤中的作用有待进一步验证。
4. 4 TRP通道家族
TRPM2 是一种可渗透Ca2 +的非选择性阳离子通道,已发现许多拮抗剂可抑制TRPM2 通道,如早期研发的氟芬那酸、益康唑和2-氨基乙氧基硼酸酯,然而因其可影响细胞内储存Ca2 +的释放而限制了临床应用。相对而言,芬那酯类似物3-MFA因其不干扰细胞内储存Ca2 + 的释放而更具有选择性。2019年的一项研究确定JNJ-28583113是一种有效的可逆的TRPM2 拮抗剂,而且其能通过血-脑屏障,但是由于其在血浆中代谢速度快,因此JNJ-28583113只能非常短暂地存在于大脑中并发挥作用,这也限制了JNJ-28583113 的进一步研究。TRPC6是经典TRPC家族的一员,正在成为控制多种疾病中Ca2 +电流的重要靶标。在一项研究中,Hyp9(一种选择性TRPC6激动剂)在体外应用时呈剂量依赖性地抑制TRPC6的下调并可减少脑缺血-再灌注损伤时星形胶质细胞的凋亡和炎性反应,在体内应用时可以减少脑梗死体积。此外,2022年的一项研究显示,TRPV3的抑制剂连翘苷B也可降低神经元过度兴奋并减轻缺血性损伤。同时TRPV4 拮抗剂HC-067047可减少小鼠脑梗死体积,而TRPV4 激动剂GSK1016790A 可通过增强NMDAR 功能等途径诱发神经毒性,促进神经元死亡。TRP通道家族众多,因此需进一步研究并设计具有特异性,且不影响细胞内储存Ca2 +释放的TRP通道抑制剂。
4. 5 线粒体对Ca2 +的摄取
MCU 与线粒体Ca2 + 内流密切相关。钌360(Ru360)是已知最有效的MCU抑制剂之一。然而,Ru360的治疗效果受其对细胞渗透性的影响。钌265(Ru265)是一种具有细胞渗透性的MCU抑制剂,其对MCU活性的抑制为Ru360的10倍,这表明只有适度增加细胞摄取才能显著改善保护作用,而且Ru265在抑制MCU的同时,可保留线粒体呼吸和糖酵解,从而改善感觉运动障碍,减少梗死体积。此外还有几种有机小分子也可作为MCU的抑制剂,如DS16570511、米托蒽醌等,但由于其明显的不良反应而限制了在体内的应用。也有研究发现,使用他莫昔芬产生条件性MCU敲除的小鼠,缺血性脑损伤诱导的感觉神经功能障碍和线粒体损伤显著减少。此外右美托咪定可通过抑制MCU,减少过度自噬,从而发挥神经保护作用。由于MCU 具有重要的作用,然而小分子作为MCU的有效抑制剂所需的结构标准尚未完善,因此需要进一步研究Ru360及其结构类似物,研制可用的高效MCU抑制剂。但是也有研究显示,MCU 的缺失显著降低了Ca2 +流入脑线粒体的速率,但并未完全消除,并且MCU的缺失在保护大脑免受缺血-再灌注损伤方面有效性有限,因此也要考虑线粒体摄取的其他途径的抑制剂。但是兰尼碱和丹曲林(均为兰尼碱受体抑制剂)也不能完全抑制线粒体对Ca2 + 的摄取,因此抑制线粒体Ca2 +摄取的途径,或许应为联合制剂,而非某一种药物能完全抑制线粒体Ca2 +超载对神经元的毒性作用。
5 展望
在缺血性脑损伤的不同阶段,[Ca2 +]i 水平变化各有其特点,并与组织病理学缺血性变化相关。细胞Ca2 +稳态丧失与缺血性脑损伤各个机制密切关联,大量动物实验或细胞实验均证实抑制Ca2 +转运体可显著抑制缺血性损伤的神经元毒性,因此,可通过血-脑屏障的Ca2 +转运体抑制剂在缺血性卒中急性期治疗中可能具有良好的神经保护作用。