河南大学生命科学学院魏建设教授团队联合郑州大学第三附属医院的乔军波教授团队在本刊发表了题为“Unraveling Parkinson's Disease: The Mystery of Mitochondria and the Role of Aging”的综述文章,第一作者为河南大学生命科学学院博士研究生刘婷婷。文章指出,健康老年人脑细胞中存在线粒体DNA(mtDNA)补偿机制,可维持线粒体功能,但帕金森病(PD)患者该机制受损,导致mtDNA减少、呼吸链功能异常及三磷酸腺苷(ATP)合成不足,神经元能量供给匮乏。同时,衰老会加剧氧化应激,破坏线粒体动态平衡,使黑质多巴胺能神经元因能量代谢紊乱而逐渐丢失。此外,衰老小胶质细胞清除神经毒性物质的能力下降,其表面触发受体2(TREM2)基因表达下调促使细胞向促炎表型转变,激活核因子κB(NF-κB)/NOD样受体热蛋白结构域相关蛋白3(NLRP3)炎症小体轴,加剧神经炎症;C-X3-C基序趋化因子配体1(CX3CL1)/C-X3-C趋化因子受体1(CX3CR1)信号通路下调则导致促炎细胞因子释放增加,加速神经元退变(图1)。
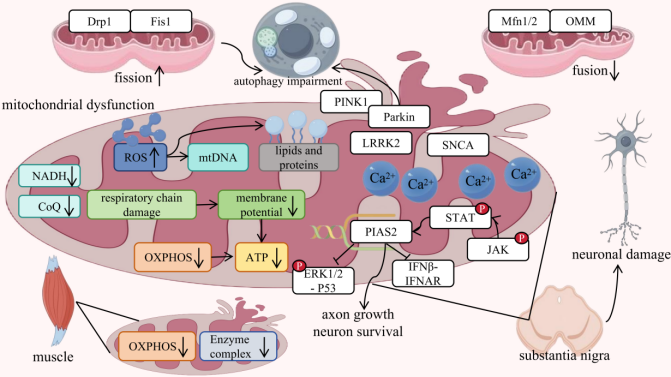
图1 帕金森病线粒体功能障碍的分子机制。(原文Figure 2)
团队进一步阐明,衰老星形胶质细胞通过环鸟苷酸-腺苷酸合成酶(cGAS)/干扰素基因刺激蛋白(STING)信号通路调控衰老进程,该通路激活后可促进促衰老因子脂质运载蛋白2(LCN2)表达,破坏星形胶质细胞代谢支持功能,加剧多巴胺能神经元丢失和运动功能障碍。在散发性PD(sPD)中,蛋白抑制剂PIAS2通过阻断Janus激酶(JAK)-信号转导和转录激活因子2(STAT2)通路,抑制线粒体自噬,导致衰老线粒体堆积和氧化应激增强,推动疾病进展(图2)。
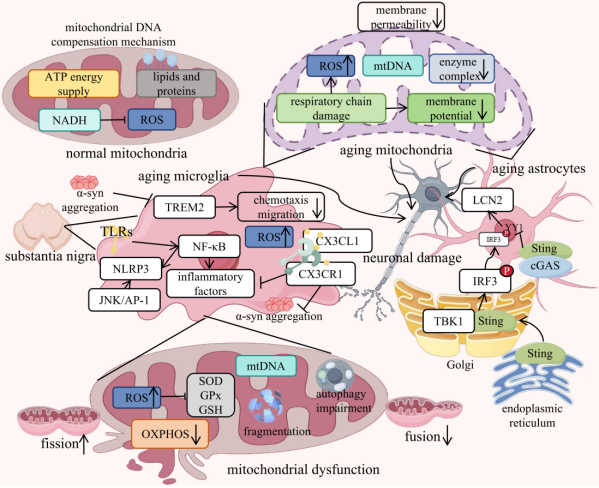
图2 衰老通过线粒体功能障碍和神经炎症加剧PD病理。(原文Figure 3)
该研究系统揭示了线粒体功能失衡、衰老及胶质细胞异常在PD中的交互作用机制,为理解PD病理进程提供了新视角。文章指出,线粒体动态失衡、自噬功能缺陷、氧化应激与钙稳态失调形成的恶性循环,是驱动多巴胺能神经元退变的核心环节,而衰老诱导的小胶质细胞和星形胶质细胞功能异常则通过重塑神经炎症微环境加速疾病发展。这些发现不仅明确了mtDNA补偿机制缺陷、cGAS/STING通路激活等关键致病节点,还提出了靶向TREM2、PIAS2及cGAS/STING轴的潜在治疗策略,为开发PD疾病修正疗法提供了重要理论依据。
免费全文下载链接:
https://www.sciencedirect.com/science/article/pii/S2352304225002089
引用这篇文章:
Liu T, Li J, Wu H, et al. Unraveling Parkinson's disease: The mystery of mitochondria and the role of aging. Genes Dis. In press.