过去五十年来,辅助生殖技术(assisted reproductive technology, ART)迅猛发展,全球已有超过1200万名婴儿借助该技术诞生。作为ART成功的关键支撑,胚胎着床前遗传学检测(preimplantation genetic testing, PGT)能够筛查染色体异常、单基因疾病及结构重排,显著提升胚胎选择准确性并降低遗传病风险。然而,传统PGT技术(如滋养层活检)仍存在操作复杂和潜在胚胎损伤风险等局限性。近年来,无创PGT(noninvasive PGT, niPGT)崭露头角,成为极具潜力的替代方案。该技术通过分析废弃胚胎培养液(spent embryo culture medium, SECM)中的游离DNA(cell-free DNA, cfDNA),大幅降低检测对胚胎的干扰。

早在2021年,北京大学第三医院乔杰/黄锦团队与北京大学生物医学前沿创新中心汤富酬/文路团队在《The Journal of Clinical Investigation》杂志上发表研究(陈依东为第一作者),将胚胎培养液中游离DNA的细胞来源追溯到囊胚细胞、颗粒细胞和极体细胞,并在此基础上精确量化了来自颗粒细胞、极体细胞的母源DNA污染水平。SECM中母源DNA的污染会导致非整倍体胚胎的检出效率显著降低,成为niPGT迈向临床应用的重要障碍之一。因此,如何优化检测方法、提高niPGT的准确性和可靠性,已成为当前生殖医学领域的重要攻关方向。
近日,北京大学第三医院乔杰/陈依东团队与北京大学生物医学前沿创新中心文路团队再次携手合作,在柳叶刀子刊《eBioMedicine》杂志发表了题为《A computational DNA methylation method to remove contaminated DNA from spent embryo culture medium for noninvasive preimplantation genetic testing》的研究论文。该研究开发了一种基于DNA甲基化信息,通过计算生物学方法降低体外培养胚胎的培养液中母源污染的新策略。这一创新性方法利用了哺乳动物胚胎发育过程中一个关键的表观遗传事件:着床前胚胎发育阶段发生的全基因组去甲基化波,该过程导致囊胚期DNA甲基化水平达到最低值。基于这一生物学现象,该研究提出通过识别并优先选择低甲基化读段来选择性富集囊胚DNA,从而将其与母源颗粒细胞DNA区分开。研究结果表明,通过筛选未甲基化读段,能够有效富集囊胚DNA并降低母源污染,这一生物信息学处理方法可显著降低存在母源污染的非整倍体SECM样本的假阴性率,有望提高无创胚胎着床前遗传检测的诊断准确性(图1)。

图1. 本图创意源于"姜太公钓鱼"的典故,巧妙隐喻低甲基化筛选技术原理。
图中姜太公手持钓竿,以低甲基化为饵(黑框白色鱼饵),成功吸引并捕获代表囊胚DNA的鱼群(左侧上钩鱼群),而代表污染DNA的其他鱼种(右侧游离鱼群)则因不喜此饵而未被捕获。这一艺术化呈现生动诠释了通过低甲基化读段筛选技术实现囊胚DNA特异性富集并有效规避污染DNA的科学机制。图中不同鱼种的差异化反应直观体现了该技术对污染DNA的筛选排除能力。
01 通过筛选低甲基化读段富集囊胚DNA
该研究首先通过分析囊胚、无颗粒细胞污染的SECM样本以及颗粒细胞的DNA在单读段水平的甲基化差异,发现三者具有显著不同的甲基化特征。基于对96例轻微颗粒细胞污染(<20%)且与TE活检CNV结果高度一致的SECM样本分析(所有样本均用单细胞全基因组甲基化建库方法,以确保可比性),结果显示:SECM/ICM/TE中未甲基化片段(0%甲基化)占比显著高于颗粒细胞(47%/50%/50% vs 12%)。通过筛选未甲基化片段可使囊胚DNA相对颗粒细胞DNA富集约4倍(47%/12%),而纳入低甲基化片段(1%-99%)虽可增加数据量但会降低富集效率至2倍(58%/26%),故最终选择未甲基化片段进行后续去污染分析(图2)。
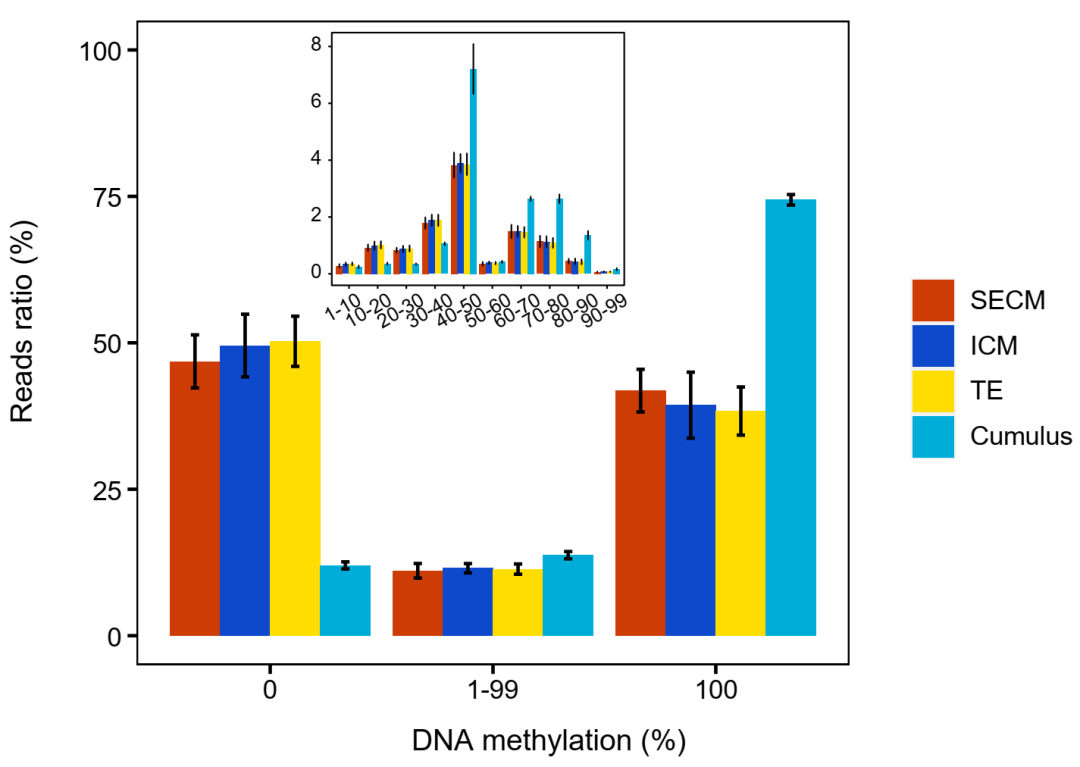
图2. SECM、ICM、TE及颗粒细胞中甲基化分布的特征分析。
X轴表示每个读段的平均DNA甲基化水平;Y轴表示在相应条件下读段的比例。其中,SECM样本为完全未受污染的培养液样本。
02 颗粒细胞污染模拟SECM样品的去污染分析
研究团队通过富集未甲基化读段,研究了从不同程度颗粒细胞污染的SECM样本中恢复囊胚DNA染色体拷贝数变异(CNV)的可行性。通过将一个非整倍体SECM样本(+22,XX,无母源污染)的读段与颗粒细胞的读段以四种不同比例(20%、50%、75%和90%)进行混合,合成了一系列模拟的DNA甲基化数据。结果显示,随着颗粒细胞污染比例的增加,非整倍体染色体的信号逐渐减弱;当颗粒细胞污染比例达到50%或更高时,非整倍体无法被检测到(图3a)。随后,研究团队筛选了未甲基化读段以排除颗粒细胞污染,并进行了非整倍体分析。结果显示,在颗粒细胞比例为50%、75%和90%的数据中,非整倍体染色体的数量明显增加,并且在颗粒细胞比例为50%和75%的数据中能够识别出非整倍体染色体(图3b)。
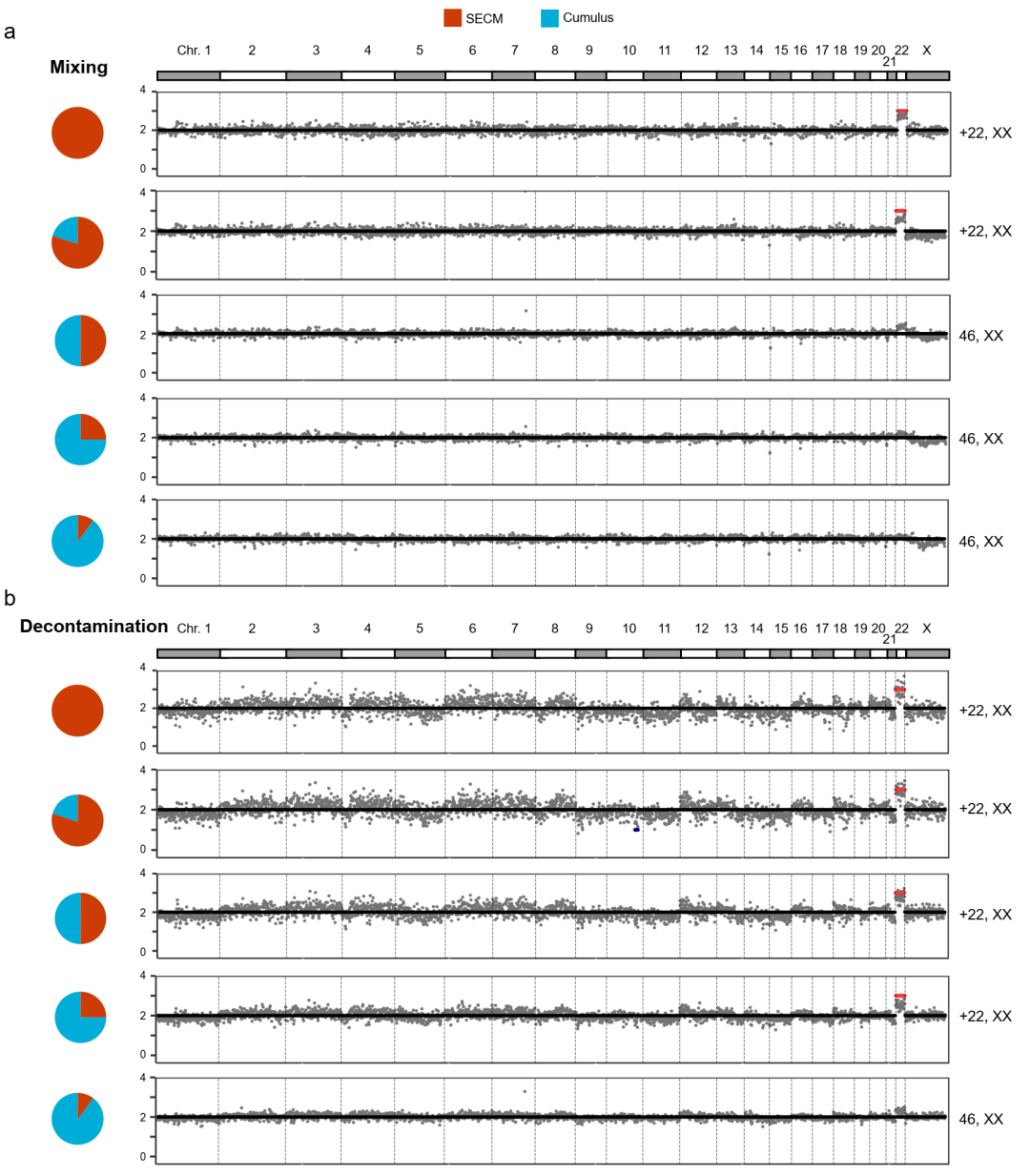
图3. 不同比例颗粒细胞污染的模拟SECM样本在去污染前后的染色体拷贝数变异(CNV)。
(a) 不同比例颗粒细胞污染的模拟样本的原始CNV。颗粒细胞的比例分别为20%、50%、75%和90%。(b) 去污染后不同比例颗粒细胞污染的模拟样本的CNV。颗粒细胞的比例分别为20%、50%、75%和90%。其中,SECM样本为完全未受污染的培养液样本。
随后,研究团队选取了所有无母源污染的非整倍体SECM样本(n=23)进行模拟分析。结果显示,虽然假阴性率始终有所降低,但部分样本显示出的CNV并非原始的非整倍体。此外,将去污染处理应用于存在母源污染的实际临床SECM样本时,也导致了较高的假阳性率。研究团队推测这些假阳性可能是由于在去污染分析过程中读段数量的减少,或是DNA甲基化区域分布的不平衡,从而引入了噪声和CNV检测的波动。为了解决高假阳性率的问题,该研究认为,当非整倍体被颗粒细胞污染掩盖时,其在去污染之前应该表现为类似嵌合体的CNV。因此,研究团队引入了一种新的算法,要求在去污染之前存在一个超过特定阈值的嵌合CNV,以及在去污染后存在一个完整的CNV。
研究团队评估了不同的去污染前CNV阈值,具体包括1.5&2.5(即拷贝数缺失的阈值为1.5,拷贝数增加的阈值为2.5)、1.6&2.4、1.7&2.3和1.8&2.2;去污染后的阈值始终设定为1.5&2.5。通过生成不同程度颗粒细胞污染的SECM样本的DNA甲基化数据,研究团队评估了通过这些不同的去污染前CNV阈值检测CNV的敏感性和特异性;每次计算实验均重复三次。
结果显示,引入去污染前的CNV阈值可以提高特异性;随着阈值从1.8&2.2增加到1.5&2.5,敏感性有所降低。在50%污染比例下,特异性从仅去污染组的87%(20/23)提高到所有整合去污染前阈值组的100%(23/23)。此外,在去污染前阈值为1.7&2.3和1.8&2.2的组别中,敏感性分别保持在83%(19/23),与仅去污染组相当。在75%污染比例下,去污染前阈值为1.8&2.2的组别与仅去污染组的敏感性相当,约为30%(7/23),而去污染前阈值为1.7&2.3的组别敏感性则降至接近零(0/23)(图4)。
上述结果表明,整合去污染前的CNV阈值1.8&2.2可以在保持与仅去污染相当的敏感性的同时,显著提高特异性。
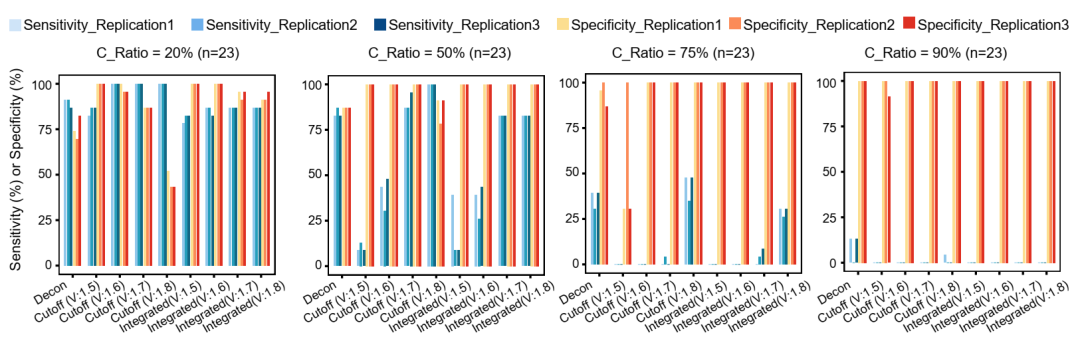
图4. 不同报出阈值下不同颗粒细胞污染SECM样品的敏感性和特异性。
03 颗粒细胞污染的临床真实SECM样品去污染分析
接下来,该研究评估了去污染算法在母源污染的实际临床SECM样本中的有效性。此前,研究者们已经报道称SECM样本通常会受到母体DNA的污染,该研究将去污染算法应用于SECM的DNA甲基化数据。去污染操作降低了假阴性率(FNR),尤其是对于母体污染比例在25%到50%之间的SECM样本。对于这些样本,仅去污染操作以及结合去污染前CNV阈值1.8&2.2的去污染操作,将FNR从原始数据中的50%降低到去污染后的21%;而当去污染前CNV阈值分别设置为1.7&2.3和1.6&2.4时,FNR分别为29%和43%。对于母源污染比例在50%到75%之间的SECM样本,原始数据、仅去污染操作以及结合去污染前CNV阈值1.8&2.2、1.7&2.3和1.6&2.4的去污染操作的FNR分别为100%、71%、86%、100%和100%(图5a)。去污染操作提高了整体的假阳性率(FPR)。对于母体污染比例在25%到50%之间的SECM样本,所有组的FPR均为40%。对于母源污染比例在50%到75%之间的SECM样本,与仅去污染操作相比,结合去污染前CNV阈值可以降低FPR。原始数据、仅去污染操作以及结合去污染前CNV阈值1.8&2.2、1.7&2.3和1.6&2.4的去污染操作的FPR分别为17%、50%、25%、25%和17%(图5b)。
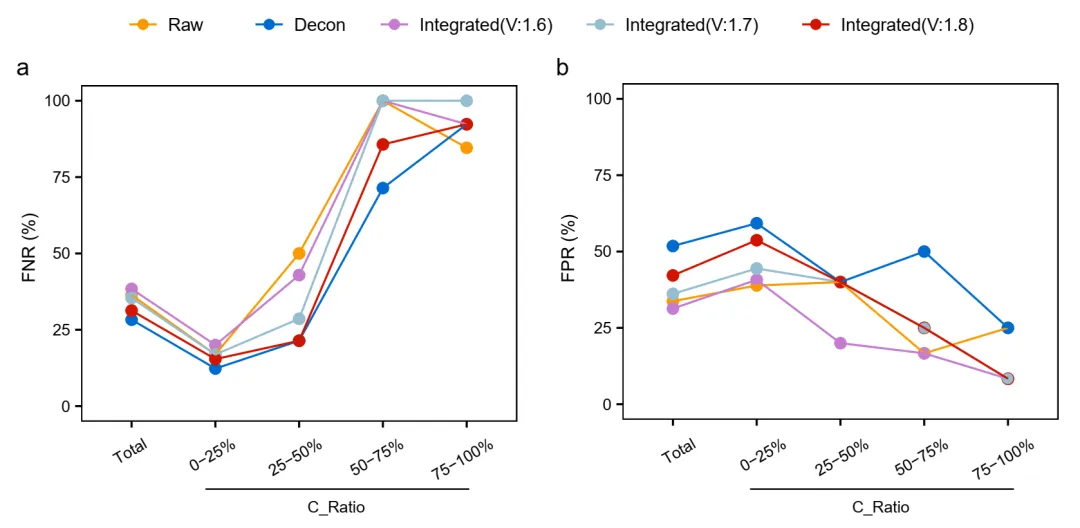
图5. 真实SECM样品去污染策略性能评估。
折线图显示不同净化策略下的假阴性率(a)和假阳性率(b)。
图6展示了三个案例。样本#S89的污染率为34.8%,初始SECM检测结果显示为整倍体状态,而相应胚胎的TE活检结果为−22,XY。经过去污染处理后,SECM检测结果被修正为−22,XY(见图6a)。样本#S28的颗粒细胞污染比例为45.5%,原始SECM的拷贝数结果为46,XX;而相应胚胎的TE活检结果为+19,XX。经过整合去污染前CNV阈值1.8&2.2的去污染操作后,SECM检测结果被修正为+19,XX,与TE活检结果一致(见图6b)。在另一个案例中,样本#S214的污染率为59.8%,原始SECM的拷贝数结果提示为整倍体,而TE活检结果为+21,XX。经过去污染处理后,SECM检测结果被正确修正为+21,XX(见图6c)。
图6. 不同颗粒细胞污染比例的真实SECM去污染整合分析前后的CNV。轻度(a)、中度(b)、高度(c)颗粒细胞污染。
04 真实临床SECM样本中非颗粒细胞DNA污染的去污染分析
进一步,研究团队探讨了选择低甲基化读段是否能够减少SECM中的精子DNA污染。在体外受精(IVF)技术应用中,精子DNA是影响胚胎外源DNA污染分析的主要干扰因素之一。与颗粒细胞相比,精子的基因组具有更高的甲基化水平,平均甲基化水平为82%(图7a)。精子中CpG位点和甲基化读段的分布呈现出独特的模式,与SECM、内细胞团(ICM)和滋养层(TE)中的模式明显不同。约65%的读段至少包含一个CpG位点,可用于读段水平的甲基化分析。其中,平均有23%、14%、8%和20%的读段分别包含1个、2个、3个或超过3个CpG位点。该研究计算了每个读段中甲基化CpG位点占所有CpG位点的百分比。对于精子样本,平均而言,20%的含CpG位点的读段甲基化值为0%,8%的读段甲基化值在1%到99%之间,72%的读段甲基化值为100%(图7b)。因此,选择未甲基化的读段可以将囊胚DNA相对于精子DNA富集约2.5倍(47%/20%)。同时,该研究使用无母源污染的整倍体SECM样本(n=23)进行模拟分析,并合成不同程度精子污染的数据。结果显示,当精子DNA占比分别为20%、50%、75%和90%时,结合去污染前阈值1.8&2.2的去污染算法的敏感性分别为87%、74%、17%和0%,特异性在96%到100%之间(图7c)。与仅去污染相比,结合去污染前阈值1.8&2.2可以提高特异性,同时在一定程度上保持敏感性。
该研究还探讨了低甲基化读段是否能够减少极体细胞的污染。由于MII卵母细胞的DNA甲基化模式与极体相似,因此使用MII卵母细胞的甲基化数据作为极体的替代。分析显示,在MII卵母细胞的甲基化数据中,38%的含CpG位点的读段未发生甲基化,这表明选择未甲基化的读段只能使囊胚DNA相对于极体DNA富集1.2倍(47%/38%;图7a和b)。通过将整倍体SECM样本(n=23)和MII卵母细胞的数据以不同比例混合进行模拟分析,结果显示,结合去污染前阈值1.8&2.2的去污染算法在极体DNA占比分别为20%、50%、75%和90%的数据中,敏感性分别为83%(19/23)、52%(12/23)、9%(2/23)和0%(0/23)(图7c)。
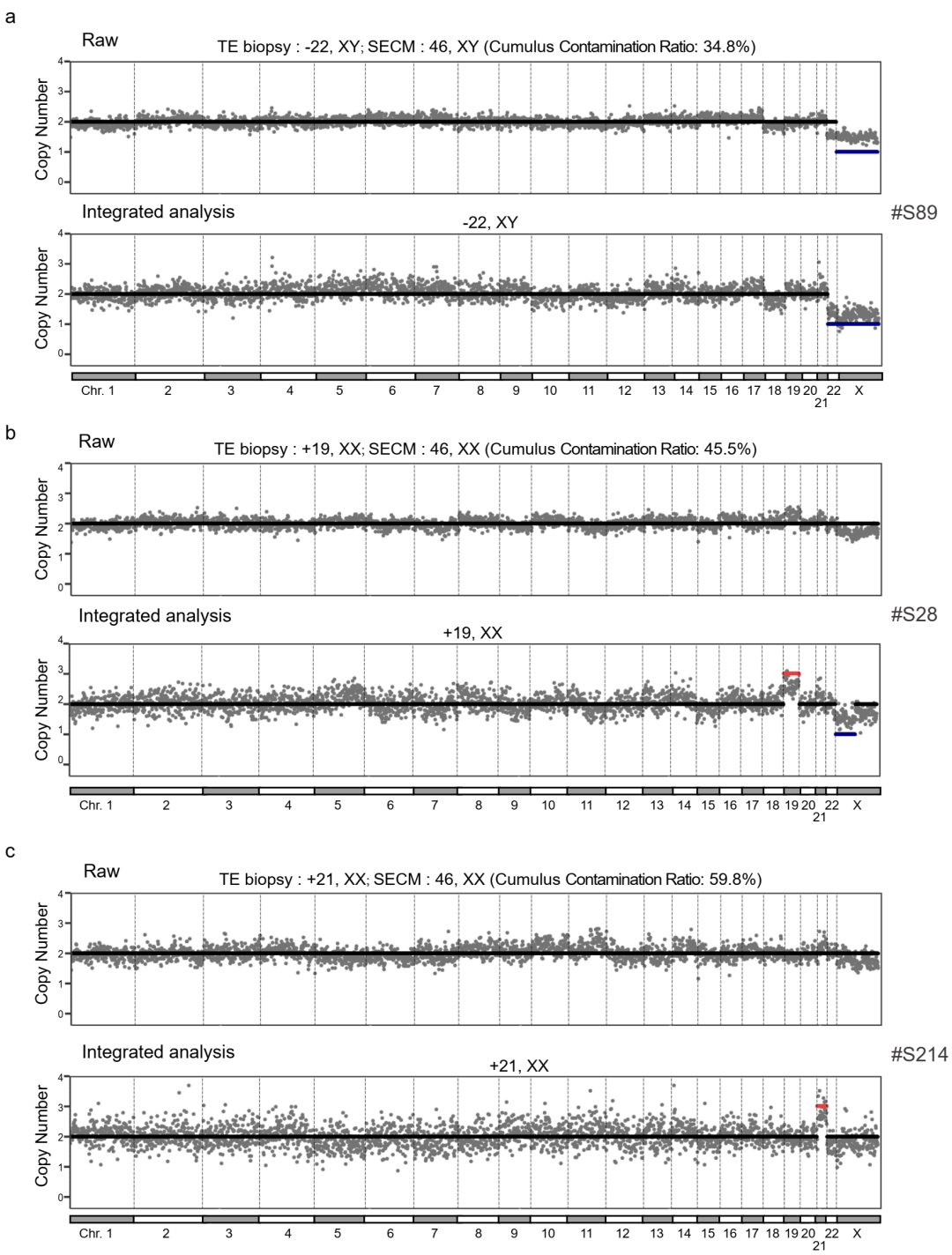
图7. 非颗粒细胞DNA污染的去污染效果评估。
(a) SECM、TE、ICM、颗粒细胞、MII卵母细胞和精子细胞的全基因组DNA甲基化水平。 (b) 左:不同CpG位点数量所占读段比例。右:每个读段的平均DNA甲基化水平分布。(c) 去污染策略在精子细胞和MII卵母细胞不同污染比例样本中的敏感性和特异性。
综上所述,该研究为减少SECM中游离DNA甲基化数据中的非胚胎污染DNA并提高非整倍体检测的敏感性提供了一种新的计算方法。结合利用DNA甲基化方法检测SECM游离DNA,该方法在无创胚胎着床前胚胎遗传学检测中具有广阔的应用前景。
北京大学第三医院陈依东副研究员为该研究论文的第一作者,北京大学第三医院乔杰院士、陈依东副研究员与北京大学生物医学前沿创新中心文路副研究员为该论文的共同通讯作者。研究团队特别感谢北京大学第三医院黄锦副主任医师和北京大学汤富酬教授的专业指导。本研究获得北京市自然科学基金、国家重点研发计划、国家自然科学基金以及中国科协青年人才托举工程等项目的资助支持。
论文链接:
https://www.sciencedirect.com/science/article/pii/S2352396425001136