近日,浙江大学良渚实验室王永成团队、浙江大学生物信息学研究所樊龙江团队、浙江大学医学院附属第一医院陈瑜团队联合M20 Genomics公司在Protein & Cell上发表题为“High-throughput single-microbe RNA sequencing reveals adaptive state heterogeneity and host-phage activity associations in human gut microbiome”的研究论文。该研究建立了基于随机引物的高通量单细胞RNA测序技术及其数据分析方法,首次实现了复杂微生物组样本的高通量单细胞转录组测序与分析。通过对人类肠道菌群单细胞转录组测序与分析,揭示了人类肠道菌群的异质性,结合针对性的计算分析方法,实现了复杂微生物群落中的微生物注释和细菌-噬菌体转录活性分析,为人类微生物组研究提供新视角。

微生物组样本的单细胞转录组测序因样本中菌群的复杂性和不同物种RNA的低捕获效率,相对纯培养样本更为困难。基于团队之前开发的smRandom-seq (Xu et al., Nat Commun, 2023),通过优化随机引物设计和反应体系,该研究开发的smRandom-seq2得以提高逆转录效率、细菌捕获效率,并有效降低交叉污染(图1)。同时,研究开发了配套的生信分析方法和工具,用于分析测序得到的微生物组单细胞转录组数据,包括单细菌注释、细菌转录表达和噬菌体转录表达三个分析模块。通过上述生信分析方法,可以实现对每个微生物的准确分类注释,并可进一步开展细菌和宿主-噬菌体关联分析。
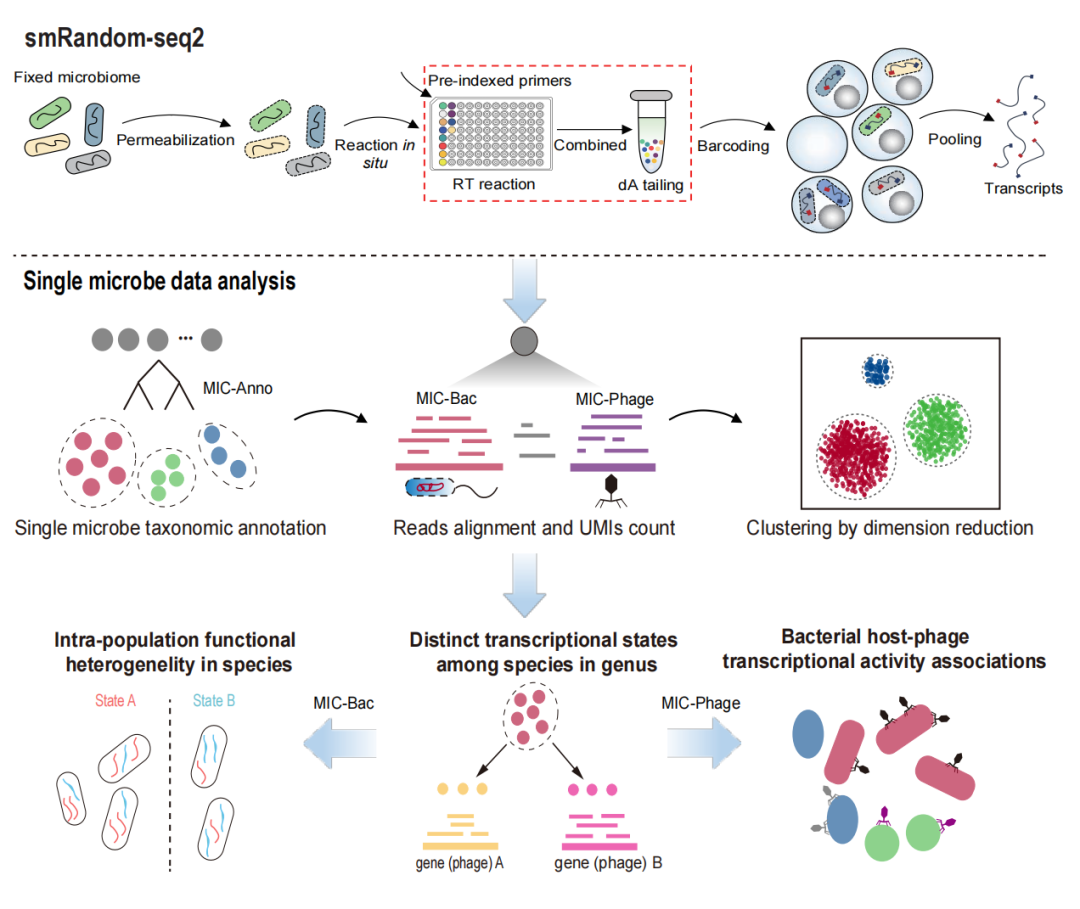
图1. smRandom-seq2工作流程示意图
该研究利用smRandom-seq2对健康人类的粪便样本进行单细胞转录组测序分析,共捕获8,478个细胞, 根据注释结果共鉴定得到98种肠道微生物,其中15个属的丰度高于1%,丰度最高的前5个属分别为Prevotella、Phascolarctobacterium、Clostridium、Dorea和Roseburia。根据肠道微生物的转录状态进行降维聚类,结果共划分出11个细菌簇,属于9个不同的细菌属,包括Prevotella、Clostridium、Fusicatenibacter、Dorea、CAG-81、Roseburia、Phascolarctobacterium、 Faecalibacterium和Lachnospira。Prevotella属和Roseburia属分别纳入了2个细菌簇,研究将这两个属的细菌簇进一步注释到物种水平。
该研究进一步分析了2种Prevotella菌之间和3种Roseburia菌之间的差异表达基因(DEG),发现同一个属的不同菌种间存在适应性反应的功能异质性。Phascolarctobacterium是肠道菌群中丰度最高的属之一,而样本中所有Phascolarctobacterium的细菌均属于同一菌种:P. succinatutens。为探索其是否存在种群内异质性,研究对其进行深入分析,聚类结果显示存在3个主要细菌亚群,并鉴定了这些亚群之间的DEG。研究发现了与可移动遗传元件以及耐药相关的基因,如ISClte1和mdtB等在亚群1中的表达水平显著提高,基因表达共现分析显示这些基因的表达之间存在显著的共现关系,提示P. succinatutens的耐药性可能来自该亚群。同时,研究发现了亚群2中琥珀酸途径的相关基因,如mutB等存在显著的高表达,提示该亚群通过琥珀酸进行化学能转换的能力更高。以上结果揭示了同一菌种在人类肠道中的功能异质性,体现了其不同的适应策略。
鉴于smRandom-seq2可以同时测定细菌及其中的噬菌体转录本,该研究建立了相应生信分析工具(MIC-Phage),在单个微生物水平上分析人类肠道菌群中宿主-噬菌体互作的转录关联。分类注释结果显示了人类肠道菌群中9个主要属的噬菌体转录谱,噬菌体和细菌转录谱的聚类结果与9个主要细菌属的匹配非常吻合,不同属间噬菌体相关基因表达水平的显著差异反映了噬菌体特异性感染的特征和能力。研究选择每个属中转录活性最高的前20个噬菌体(共180个噬菌体)进一步分析,鉴定得到373种可靠的宿主-噬菌体关联,其中至少有325种为新发现,体现smRandom-seq2在建立准确的体内宿主-噬菌体转录活性连关联方面的优势。
该研究开发的高通量单细胞转录组测序技术smRandom-seq2,克服了以往技术的种种限制,能够处理复杂的微生物群落样本,可在单个细菌的分辨率上高效率、高灵敏地分析各种微生物菌群。利用这一技术捕获复杂微生物群落中细菌的功能异质性,本研究揭示了人肠道菌群中细菌属内、种内的适应性策略异质性,发现了数百个新的宿主-噬菌体转录活性关联,开启了人类微生物组研究的新方向。
浙江大学医学院附属第一医院沈一飞副研究员、浙江大学农业与生物技术学院生物信息学研究所博士生钱青宏、浙江大学良渚实验室博士后丁利国、浙一医院硕士生瞿闻馨、M20 Genomics张天宇和宋孟迪为共同第一作者,王永成研究员和樊龙江教授为共同通讯作者。
论文原文:
Yifei Shen, Qinghong Qian, Liguo Ding, et al. High-throughput single-microbe RNA sequencing reveals adaptive state heterogeneity and host-phage activity associations in human gut microbiome. Protein & Cell, pwae027, https://doi.org/10.1093/procel/pwae027