在病理年会正式开幕前,共开展了三期“读片会会前会”。病理年会特辑第三期将3月13日读片会中,空军军医大学附属西京医院病理科克祯彧老师主讲的内容“先天性肌病伴脊柱侧弯1例”整理提炼分享,供大家随时查看学习。

内容详情
一、病例特征
14岁女性,2022年发现脊柱侧弯4年,加重伴呼吸困难半年。
病史:2018年发现双肩不等高,X射线显示脊柱侧弯,胸弯为著,Cobbs角度21.8°,后矫形治疗至9.1°。患儿未规律佩戴支具,无规律复查,半年前症状加重,伴呼吸困难,Cobbs角增至90.6°。
体征:身高140cm,体重22kg,触诊棘突呈左凹右凸S样畸形,按压痛(-),叩击痛(-)。剃刀背畸形,四肢纤细,肌肉严重萎缩,肌肉容量低,下蹲后无法自行站立。双上肢肌力尚可,双下肢肌力下降。
辅助检查:肺功能显示极重度限制性通气功能障碍,右肺压缩严重。心电图和心脏彩超未见异常。牵引45天后与牵引前变化不大。其他血清学检查均正常。
二、多学科诊疗(MDT)
1.肌肉活检:患儿四肢非常纤细,肌肉严重萎缩,肌肉容量低下,蹲后无法自行站立,下肢的肌力也明显下降,考虑脊柱侧弯为肌肉源性,需肌肉活检进一步明确。
2.加强营养
3.头环牵引
4.呼吸功能锻炼
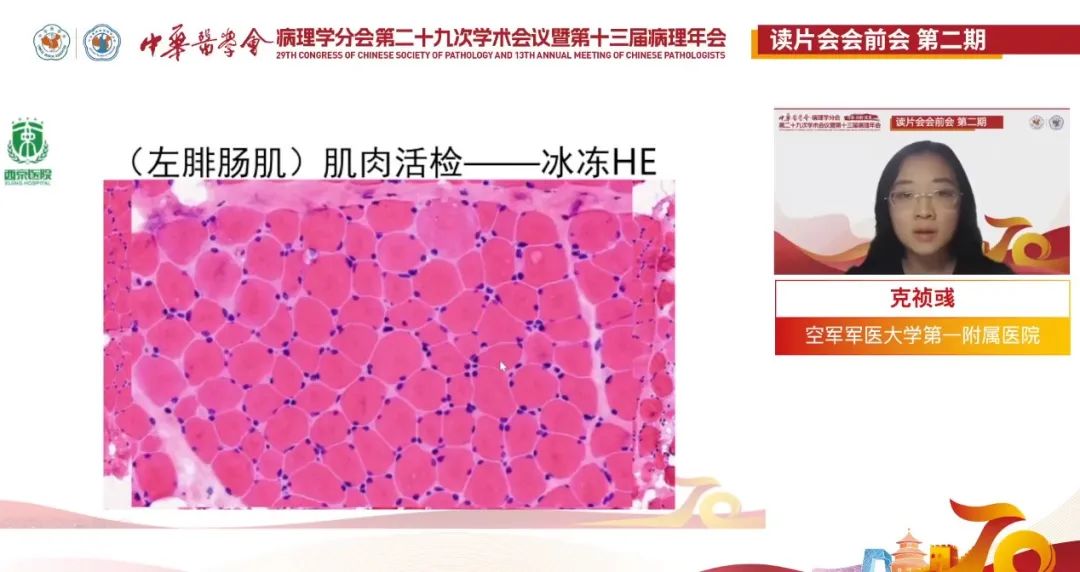
(左腓肠肌)肌肉活检--冰冻HE:
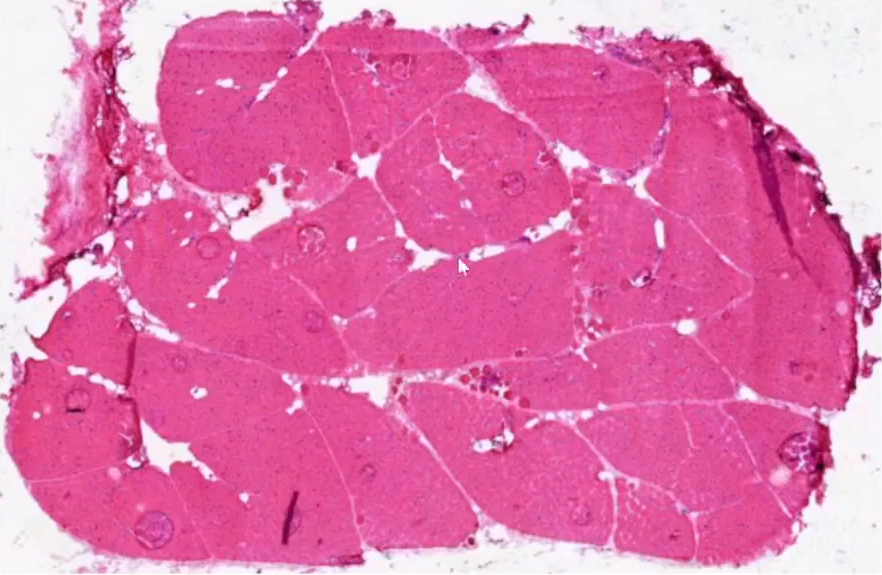
低倍镜下:肌纤维萎缩,肌间脂肪增多。
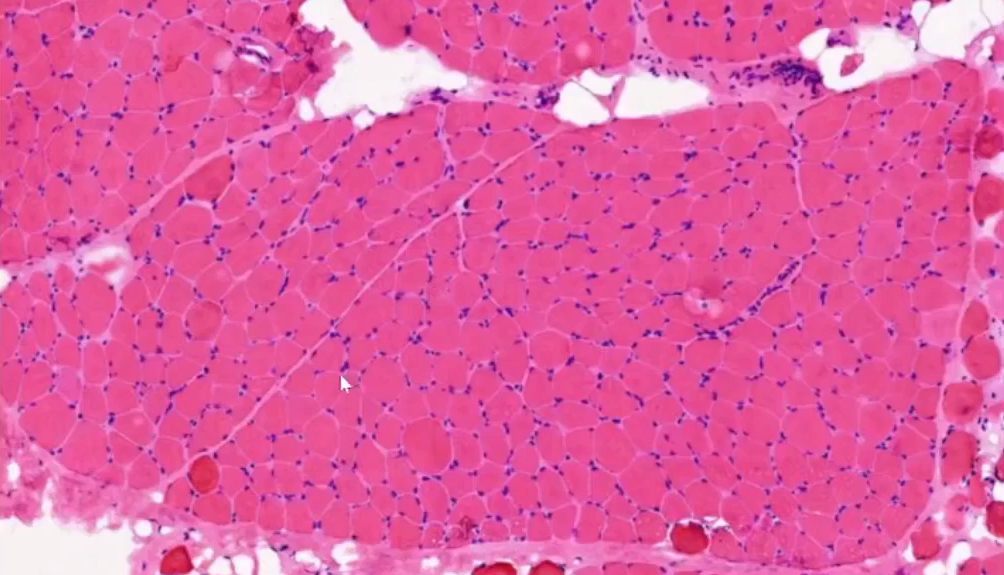
放大:肌纤维粗细不等,部分显著萎缩,部分肌纤维肥大圆形化(正常肌纤维多角形)。
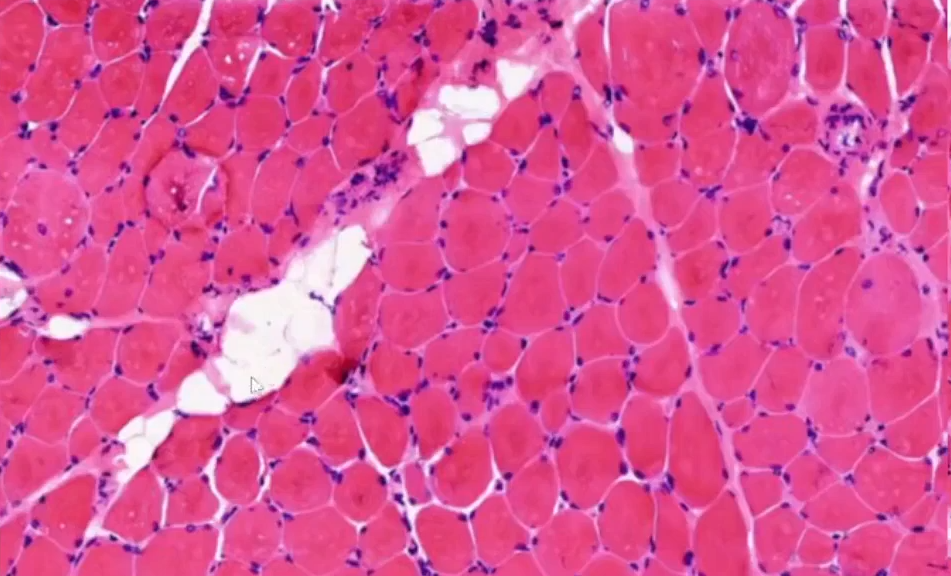
再放大:部分肌纤维变性,间质纤维组织及脂肪组织增生,伴有少量炎细胞浸润,另外观察到少许细胞出现核内移现象,肌纤维中心结构异常不明显。
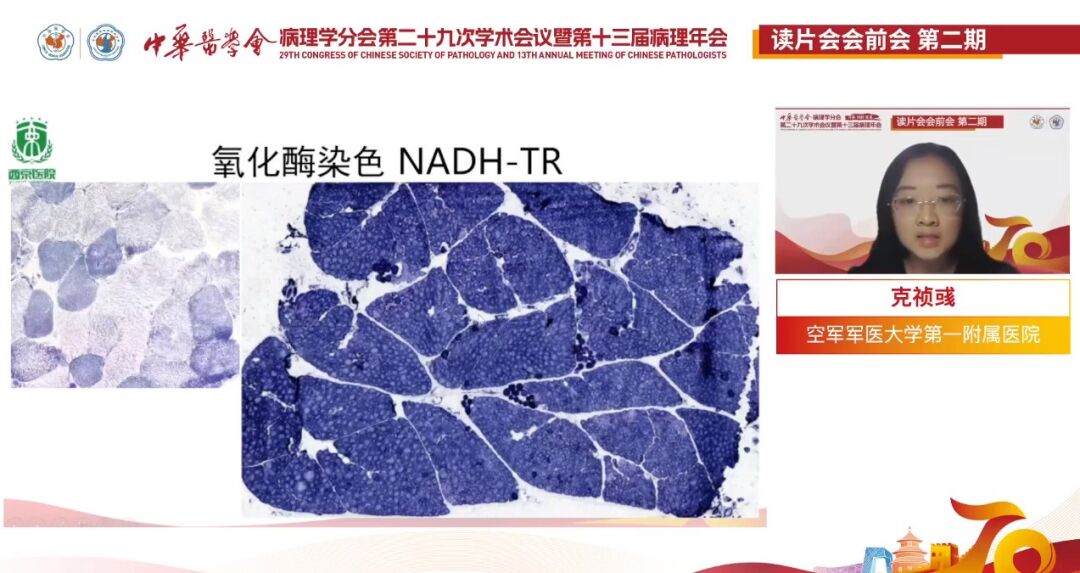
氧化酶染色(NADH-TR):
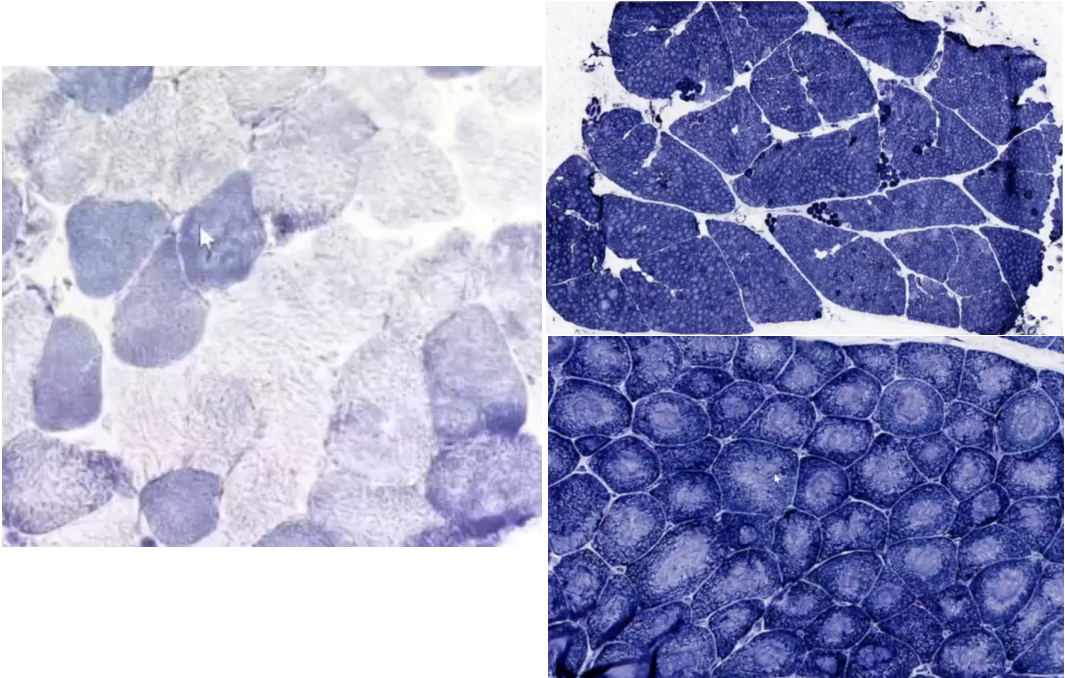
患儿(右):显示Ⅰ型肌纤维中央广泛空洞形成(右上);以Ⅰ型肌纤维为主,且中央结构淡染(右下),Ⅱ型几乎不可见。正常肌纤维(左):深染以Ⅰ型肌纤维为主,淡染为ⅡA、ⅡB型,正常情况下Ⅰ型和Ⅱ型镶嵌分布。
电镜:

肌原纤维z线显著紊乱,呈水管状或蛇形排列(正常z线为平行排列)。
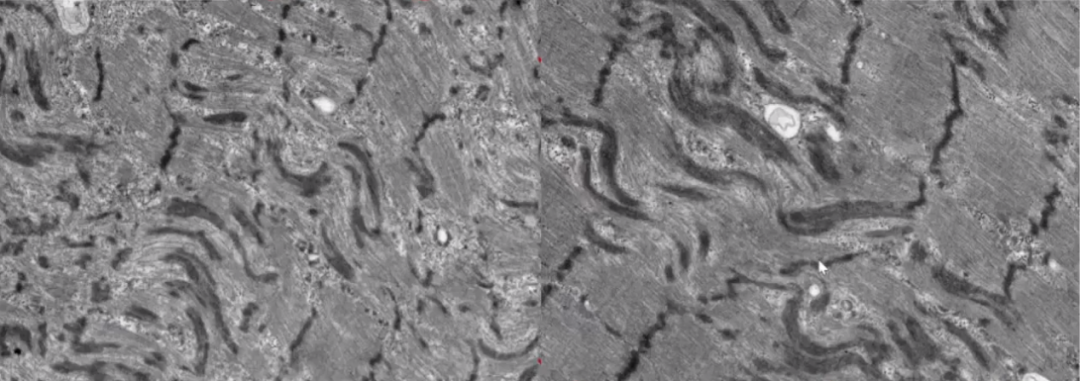
放大后可见显著紊乱的z线,未见线粒体。
肌肉活检结果:
形态学考虑中央轴空病,建议临床行相关分子检测进一步证实。
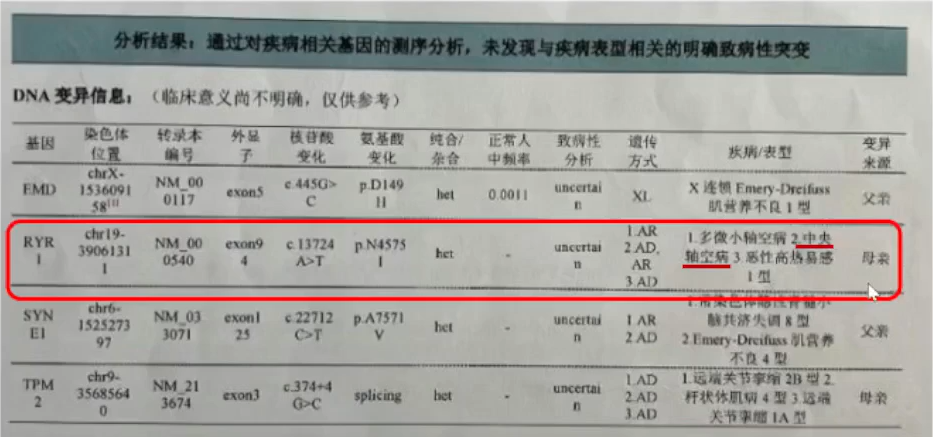
基因检测:
检测到来自母亲的RYR1基因杂合性突变,该基因多见于中央轴空病。母亲身材矮小(身高1.4米),有肌无力表现。父亲体型正常(身高1.7米),身体健康。基因检测证实患儿的基因异常来自母亲。
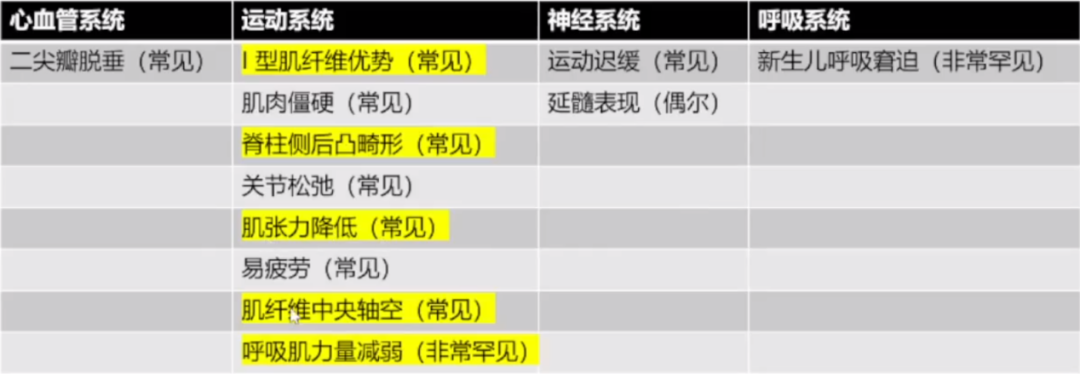
三、中央轴空病:
中央轴空病(Central core dⅠsease,CCD)是一种轻度到重度肌无力疾病,1956年由Shy和Magee首次报道,多为常染色体显性遗传,部分病例为常染色体隐性遗传,致病基因RYR1,编码斯里兰卡肉桂碱受体1蛋白,该蛋白位于骨骼肌肌质网,组成钙离子释放通道,在骨骼肌的兴奋收缩耦联中发挥重要作用。
临床症状:
出生时起病,主要表现为近端肌无力、肌张力低下,一般下肢较上肢重,近端比远端重,腱反射减弱或消失,可伴运动发育迟缓,常伴先天性髋关节脱位、脊柱侧弯、关节挛缩、弓形足等,可有轻度的颜面肌和颈肌受累。
CK多正常或轻度升高,EMG示肌源性和/或神经源性损害。
病程为非进展性或缓慢进展。一般能够独立行走、寿命正常。
典型病理表现:
NADH-TR等氧化酶染色可见肌纤维胞浆中有类圆形不着色区——空白的水果核样结构,而HE、MGT染色,肌纤维内轴空结构不明显。
此外,可见肌纤维大小不一,呈圆形化,结缔组织增生等,变性、坏死肌纤维少见,多数病例Ⅰ型肌纤维优势。
电镜下,中央轴空区域线粒体、肌质网一般减少或消失,肌原纤维结构素乱,Z线呈蛇行状或水纹状,部分患者可见轴空区由变异的线粒体和大量糖原颗粒填充。
CCD的诊断主要依靠肌肉活检及患者的临床表现。
典型患者病理活检显示轴空位于Ⅰ型肌纤维的中央,但轴空的数量与疾病的严重程度暂时未发现具有显著相关性。在发病的早期阶段,少数患儿病理上可以无轴空结构出现。CCD需与微小轴空病等先天性肌病鉴别。

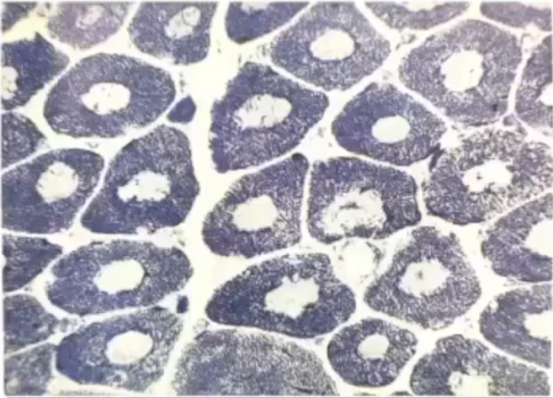
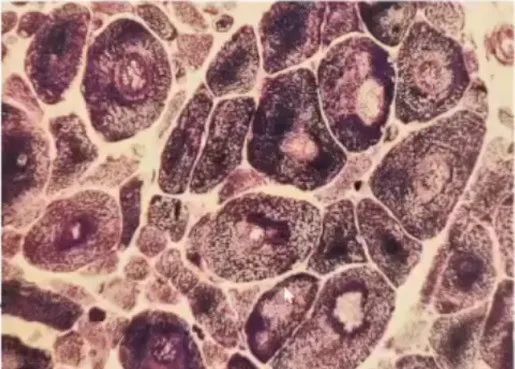
左图:中央轴空病(轴空结构)镜下形态;右图:肌萎缩侧索硬化ALS(靶状纤维)镜下形态
四、治疗与预后
经6次多学科讨论后最终制定方案:
-
术前Halo头环牵引-呼吸功能锻炼-营养支持。
-
术中预防恶性高热-预备特效药物(丹曲林--肌松剂)-快速有效矫形。
-
术后重症医学监护-评估呼吸功能-功能训练。
经过长达三个月的术前准备后,顺利手术,术后呼吸功能恢复正常,脊柱侧弯矫正满意。患者术后恢复迅速,一个月后出院,定期复查无问题。
病例启示:强调肌肉活检的重要性和必要性,对明确诊断有极大帮助。